
Rare combination of dilated cardiomyopathy and ankylosing spondylitis in a family
2018-10-15 04:03:46YunLiXINGYingSUNQingMAYuLUYingSHAOWeiHUANGDaiZHANGFuShengGUHongWeiLI
Journal of Geriatric Cardiology 2018年8期
Yun-Li XING, Ying SUN, Qing MA, Yu LU, Ying SHAO, Wei HUANG, Dai ZHANG, Fu-Sheng GU, Hong-Wei LI
?
Rare combination of dilated cardiomyopathy and ankylosing spondylitis in a family
Yun-Li XING, Ying SUN, Qing MA, Yu LU, Ying SHAO, Wei HUANG, Dai ZHANG, Fu-Sheng GU*, Hong-Wei LI*
Department of Geriatrics and Gerontology, Beijing Friendship Hospital, Capital Medical University, Beijing, China
J Geriatr Cardiol 2018; 15: 554?556. doi:10.11909/j.issn.1671-5411.2018.08.002
Ankylosing spondylitis; Familial dilated cardiomyopathy; Genetic mutation
A 65-year-old man presented to our cardiovascular department due to fatigue and palpation on exertion during the previous three weeks. He had a medical history of diabetes mellitus and hyperlipemia without hypertension or myocarditis. However, he mentioned that his son had dilated cardiomyopathy (DCM) and ankylosing spondylitis (AS). Examination at admission revealed a blood pressure of 115/69 mmHg and pulse of 82 beats/min. The results of routine blood tests for creatine kinase (CK), CK muscle and brain (CK-MB), troponin T, and thyroid function were all within normal limits. ECG revealed I, aVL, and V4–6 T wave inversion (Figure 1). Ultrasonic cardiography showed that the left ventricular ejection fraction (EF) was 32%, indicating ventricular wall dysfunction. Angiography showed no coronary stenosis. Radionuclide imaging demonstrated myopathy of the anterior, part of the inferior, the posterior, and the lateral apical segments based on reduced radioactivity with a patchy pattern, consistent with DCM (Figure 2A). Cardiac magnetic resonance imaging confirmed the decreased left ventricular motion and late gadolinium enhancement in the septal wall (Figure 2B), confirming the diagnosis of DCM.[1]Based on the patient’s family history of AS, we performed computed tomography examination of the sacroiliac joint and tested for human leukocyte antigen B27 (HLA-B27). Observation of the fused joint and the positive result for HLA-B27 supported the diagnosis of AS. However, we needed to discriminate the familial DCM from AS-induced myocardiopathy. The patient’s mother had experienced sudden cardiac death a few years previously. Among the five children of his older aunt, one had DCM and one had AS, and the two grandchildren of this aunt had AS. Among the six children of his younger aunt, one had AS and one had both AS and DCM. Also, one grandchild of this aunt had AS. The patient’s family pedigree is shown in Figure 3. His own granddaughter had no dyspnea or palpitation, and ECG was normal. The strongly suspected familial DCM was confirmed by gene expression analysis includingthe patient, his son and his granddaughter, which showed variations in the titin (TTN) and Bcl-2 associated anthanogene-3 (BAG3) genes in the father and son, but not in the granddaughter, consistent with the clinical manifestations (Figure 4). The patient was treated with medications including the angiotensin receptor blocker, metoprolol 25 mg (t.i.d.), furosemide 40 mg (q.d.), aldactone 20 mg (b.i.d.), and ivabradine 5 mg (b.i.d.), along with cardiac rehabilitation. After six months, he could run on a treadmill for 1 h without dyspnea, and his cardiac function had improved with an EF ranging from 40%–51%.
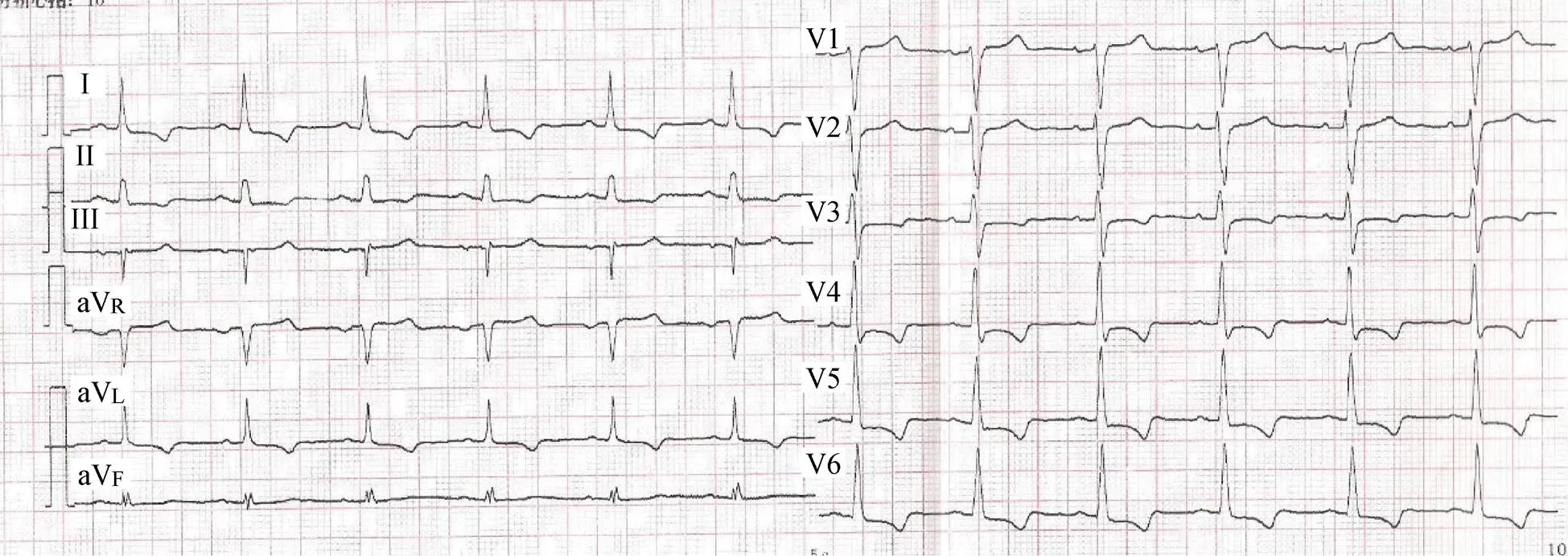
Figure 1. ECG demonstrating I, aVL, and V4–6 T wave inversion.
Figure 2. Radionuclide imaging (99mTC-MIBI) and cardiac magnetic resonance imaging. (A): Radionuclide imaging (99mTC-MIBI): myopathy of the anterior, part of the inferior, posterior, and lateral apical segments with reduced radio activity. (B): Cardiac magnetic resonance: long axis images (a–b) demonstrated left ventricle (LV) dilation and reduced systolic function. Short axis images (c–d): delayed gadolinium enhancement in the septal wall is noted in c (arrow).99mTC-MIBI: 99 m technetium-methoxyisobutylisonitrile.
To the best of our knowledge, few familial cases of both DCM and AS have been reported. Notably, autoimmunity plays a vital role in myocarditis and DCM, and research has linked HLA status with a predisposition to familial DCM.[2]Several challenges for DCM management, such as the interpretation of genetic testing results, the correct treatment for pre-clinical asymptomatic DCM gene carriers, and the development of gene- and mechanism-specific therapies.[3]
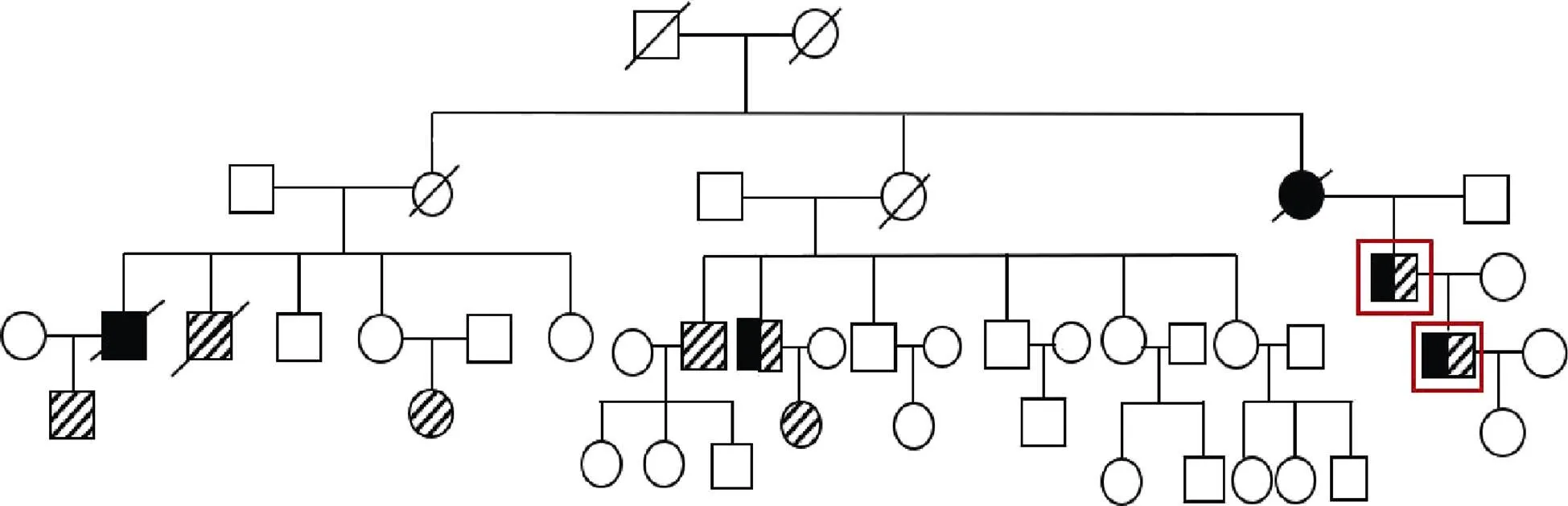
Figure 3. Pedigree of the patient’s family showing diagnoses of DCM and/or AS. Squares indicate men; circles, women; crossed symbols, death; black highlighted symbols, diagnosed with DCM; slash highlighted symbols, diagnosed with AS; red squares, father and son; clear symbol, number of unaffected members. AS: ankylosing spondylitis; DCM: dilated cardiomyopathy.
Figure 4. TTN and BAG3 gene expression of the father, son and granddaughter. Father and son showed TTN (left) and BAG3 (right) gene variation. Red arrows indicated variation position. BAG3: Bcl-2 associated anthanogene-3; TTN: Titin.
1 Japp AG, Gulati A, Cook SA,. The diagnosis and evaluation of dilated cardiomyopathy.2016; 67: 2996–3010.
2 McKenna CJ, Codd MB, McCann HA,. Idiopathic dilated cardiomyopathy: familial prevalence and HLA distri-bution.1997; 77: 549–552.
3 McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms.2017; 121: 731–748.
Correspondence to:gufusheng314@sina.com (GU FS); lhw19656@sina.com (LI HW)
 Journal of Geriatric Cardiology2018年8期
Journal of Geriatric Cardiology2018年8期
- Journal of Geriatric Cardiology的其它文章
- Patent foramen ovale in an old patient with ischemic stroke following carotid surgery
- Normalization of plasma growth hormone alleviated malignant ventricular tachycardia in acromegaly
- Natriuretic peptide family as diagnostic/prognostic biomarker and treatment modality in management of adult and geriatric patients with heart failure: remaining issues and challenges
- Subclinical hypothyroidism is associated with lipid-rich plaques in patients with coronary artery disease as assessed by optical coherence tomography
- Five-year major clinical outcomes between first-generation and second- generation drug-eluting stents in acute myocardial infarction patients underwent percutaneous coronary intervention
- Correction of hypovitaminosis D improved global longitudinal strain earlier than left ventricular ejection fraction in cardiovascular older adults after orthopaedic surgery