
The first draft genome assembly and data analysis of the Malaysian mahseer(Tor tambroides)
2023-10-20 01:45:50MelindMeiLinLuLeonrdWhyeKitLimHungHuiChungHnMingGn
Aquaculture and Fisheries 2023年5期
Melind Mei Lin Lu, Leonrd Whye Kit Lim, Hung Hui Chung,*, Hn Ming Gn
a Faculty of Resource Science and Technology, Universiti Malaysia Sarawak, Kota Samarahan, Sarawak, 94300, Malaysia
b GeneSEQ Sdn Bhd, Bandar Bukit Beruntung, Rawang, Selangor, 48300, Malaysia
c Centre for Integrative Ecology, School of Life and Environmental Sciences, Deakin University, Geelong, Victoria, VIC3220, Australia
Keywords:Genome Gene annotation Tor tambroides Phylogenetic Functional annotation
A B S T R A C T The Malaysian mahseer (Tor tambroides), one of the most valuable freshwater fish in the world, is mainly targeted for human consumption.The mitogenomic data of this species is available to date, but the genomic information is still lacking.For the first time, we sequenced the whole genome of an adult fish on both Illumina and Nanopore platforms.The hybrid genome assembly had resulted in a sum of 1.23 Gb genomic sequence from the 44,726 contigs found with 44 kb N50 length and BUSCO genome completeness of 87.6%.Four types of SSRs had been detected and identified within the genome with a greater AT abundance than that of GC.Predicted protein sequences had been functionally annotated to public databases, namely GO, KEGG and COG.A maximum likelihood phylogenomic tree containing 52 Actinopterygii species and one Sarcopterygii species as outgroup was constructed, providing first insights into the genome-based evolutionary relationship of T. tambroides with other ray-finned fish.These data are crucial in facilitating the study of population genomics, species identification,morphological variations, and evolutionary biology, which are helpful in the conservation of this species.
1.Introduction
The Malaysian mahseer,Tor tambroides(Bleeker, 1854), one of the members of the family Cyprinidae, is a widespread species found in aquaculture and fisheries mainly targeted for human consumption(Kottelat et al.,2018).It is commonly named Kelah or Empurau in Malaysia and Jurung, Indonesia (Jaafar et al., 2021).As a true mahseer(Torspp.), it can be found in rapidly-flowing waters with rocky bottoms(Shreshtha, 1997).Together withTor tambraandTor dourenensis,it is one of the threeTorspp.found in freshwaters of Malaysia, and among the 16Torspp.found worldwide (Ng, 2004).
Like otherTorspp.,T.tambroidesis endangered by environmental degradation within their habitat, causing an elevating shrink in their population size in recent years (Ingram et al., 2005).Anthropogenic modification of rivers, including agricultural activities, logging, and deforestations, had interrupted and reduced the water flow within its habitat.Consequently, such actions not only shrink the population size ofT.tambroidesand other Tor spp., but it is also associated with impacts on the aquatic environment.Despite environmental issues,T.tambroidesare also at threat from overfishing with the usage of hooks, nets, and dynamites.However, there is currently no information on the rate of these losses (Kottelat et al., 2018).
It resides a wide range of freshwaters, including Brunei Darussalam,Yunnan China, Indonesia (Kalimantan, Jawa, and Sumatera), Laos,Malaysia (Peninsular, Sabah, and Sarawak) and Thailand (Kottelat,2000).Despite its comprehensive habitat coverage, its distant population can probably be different, leaving its taxonomy still vague.Its body is covered with scales depending on locality, such as silver, bronze, and reddish.
The accumulation of significant genetic and/or morphological differences amongTorspp distributed across Indonesia and Malaysia urged the revision ofTortaxonomy.Studies had been done aiming to resolve the ambiguous taxonomical status ofTorspp.by looking into its phylogenetic relationship through mitochondrial DNA, CytochromecOxidase Subunit I (COX1) gene, Cytochromeb, ATPase 6/8 gene, 16S rRNA gene, microsatellite, and SNP markers (Walton, Gan, Raghavan,Pinder, & Ahmad, 2017; Jaafar et al., 2021; Lim, Chung, Lau, et al.,2021).However, to date, the whole genome and transcriptome sequences ofTorspp.are still unavailable, except for a few studies that had reviewed its conservation status, conducted a gut metagenomic analysis,as well as sequenced the transcriptome ofT.tambra(Lau et al., 2021a,2021b, 2021c).It had been reported earlier thatT.tambroidesis a tetraploid, having 2n =100 chromosomes (Donsakul & Magtoon, 2008).Genomic and transcriptomic sequencing ofTorspecies is necessary to provide a more powerful tool that continues to resolve and address questions of species identification, evolutionary biology, morphological variations, sequences related to sex differentiation, growth, reproduction, and immune, which is helpful for further conservation ofTorspecies (Jaafar et al., 2021).
The rapid growth and expansion of the aquaculture industry had improved fish production as compared to traditional fishing.T.tambroidesis one of the notable species found in the aquaculture industry due to its high nutritional value and unique flesh taste.However,it has been found that the genetic variability decreases in captivity-held fishes, causing them to be more vulnerable to infections.Thus, it is necessary to comprehend the fish immune system and its underlying mechanisms in response to its exposure to ecotoxicological chemicals(Lim et al., 2018, 2021b).However, in general, the studies of the fish immune response are limited due to its complexity and lack of suitable reagents for classical immunological assays (Salinas & Magadán, 2017).Similarly, as a slow grower fish, it is necessary to venture into the growth-related aspect of theT.tambroidesgenome as well, which is in line with the goal of fish farming of this species which faces knowledge-scarcity on growth improvement to date.Omics approaches,including genomics, transcriptomics, proteomics, and metabolomics,can develop high throughput outcomes and facilitate more novel findings.
In the recent years, with the booming of long-read sequencing technologies (Heather & Chain, 2016), the implementation of combining both Illumina reads and Nanopore/PacBio reads can be found in several studies (Austin et al., 2017; Lim et al., 2021c, 2022; Tan et al., 2018).Integration of short but accurate Illumina reads with long but less accurate Nanopore/PacBio reads could enhance the completeness of the assembled genome than assemblies based on Illumina reads only (Austin et al., 2017; Lim et al., 2021c, 2022; Tan et al., 2018).Thus,in this study, we sequenced and reported on the first genomic data ofT.tambroides.We also functionally annotated the genome ofT.tambroidescontaining 76,419 predicted non-redundant protein sequences to KEGG, GO, and COG databases and further identified its immune-related genes.Furthermore, we inferred the phylogenetic relationship ofT.tambroideswith other Actinopterygii fishes based on the BUSCO supermatrix data.It is hoped that these data generated from this study can be channeled for the improvement studies that are driven along with the conservation endeavors of this fish species.
2.Experimental design, materials and methods
2.1. Ethics statement
All experiments comply with ARRIVE guidelines and were carried out in accordance with the U.K.Animals (Scientific Procedures) Act,1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments, or the National Institute of Health guide for the care and use of Laboratory animals (NIH Publications No.8023, revised 1978).
2.2. Sampling and DNA extraction
An adult maleT.tambroides(voucher ID: ASD03018) was sampled from a local aquaculture farm, with its locality reported in previous studies (Lau et al., 2021c; Lei et al., 2021).The fish had been deposited as voucher specimens in the fish museum located at the Faculty of Resource Science and Technology, Universiti Malaysia Sarawak.The fish was euthanised, and 50 mg of its muscle tissue was used for genomic DNA extraction using the DTAB-CTAB DNA extraction kit (GeneReach Biotechnology Corp) according to the manufacturer’s instructions.
2.3. Species verification
Cytochromecoxidase subunit I (COI) gene was amplified, and the PCR product was purified and sent for Sanger sequencing.The sequences were analysed using NCBI BLASTn and found to possess 100% similarity with the mitogenome sequences ofT.tambroidesreported by Lei et al.(2021) with the GenBank accession number MW471071.1.
2.4. Library construction and whole genome sequencing
Approximately one ug of gDNA was sheared to 350 bp using a Bioruptor and directly used for PCR-free library preparation using the NEB Ultra Illumina library preparation kit (NEB, Ipswich, MA).The library was quantified with a Qubit (Invitrogen) and sequenced on a Nova-SEQ6000 (Illumina, San Diego, CA) with 2 ×150 bp run configuration.Similarly, for Nanopore sequencing, one ug of unsheared gDNA was used as the input for LSK109 library preparation (Oxford Nanopore, UK)according to the manufacturer’s instructions.The library was sequenced on two MinION flowcells.Nanopore reads were base-called from their fast5 files using Guppy version 4.4.1 (high accuracy mode).
2.5. Sequence data processing and assembly
Illumina reads were trimmed with fastp (Chen et al., 2018), while the Nanopore reads were trimmed with porechop (Wick et al., 2017).The trimmed Nanopore reads and paired-end Illumina reads were used to perform a hybridde novoassembly using Wengan v0.2 (Di Genova et al.,2021).The genome assembly was subsequently polished with nextpolish(Hu et al., 2020) based on alignment of Illumina reads to the genome.Jellyfish v2.3.0 was used to obtain a frequency distribution of k-mer counting with the clean reads, producing kmer frequency distributions of 31-mers (Mar?ais & Kingsford, 2011).Clean Illumina reads were subsequently aligned to the polished genome using Bowtie2 to assess genome mapping rate.(Langmead & Salzberg, 2012).These histograms were subsequently processed using GenomeScope, which estimates genomic size, repeat content and heterozygosity via kmer-based statistical approach (Vurture et al., 2017).QUAST v5.0.2 was used to evaluate various metrics of theT.tambroidesgenome (Mikheenko et al., 2018)using the parameter: read length 150 bp and max K-mer coverage 1000.BUSCO v5.3.0 was used to evaluate the completeness of the assembledT.tambroidesgenome and other publicly available fish genomes based on the single-copy orthologs represented in the actinopterygii_odb10 database (Manni et al., 2021).
2.6. Detection of repetitive sequences and prediction of protein-coding genes
Repetitive sequences in the assembled genomes were identified and masked using RepeatModeler2 and RepeatMasker, respectively.Previously generated transcriptomic reads (Lau et al., 2021b) were aligned to the repeat-masked genome assembly using HiSAT2 (Kim et al., 2015).The transcriptome alignment BAM file and the repeat-masked genome assembly were used as the input for protein-coding gene prediction in BRAKER1 (Bruna et al., 2021).An additional annotation was also performed via the alignment of vertebrate odb10 conserved proteins using BRAKER2 (Bruna et al., 2021).Both BRAKER1 and BRAKER2 annotations were combined and filtered with TSEBRA (Gabriel et al., 2021),generating a final set of predicted protein-coding genes.
2.7. SSR analysis
Simple sequence repeats (SSRs) analysis was identified using Kmer-SSR (https://github.com/ridgelab/Kmer-SSR) (Pickett et al., 2017) on the processed reads of theT.tambroidesgenome.Dinucleotides, trinucleotides, tetranucleotides, pentanucleotides and hexanucleotides were included in the SSRs analysis.Only SSRs with at least four repeats were selected for the study.
2.8. Functional annotation of protein-coding genes
The predicted protein sequences were functionally annotated to EggNOG mapper (evolutionary genealogy of genes: Non-supervised Orthologous Groups) with a minimum E-value of 0.001.Functional annotation of genes was performed by mapping against three public databases, GO (Gene ontology), KEGG (Kyoto Encyclopedia of Genes and Genomes), and COG (the Clusters of Orthologous Groups).
In addition, predicted protein sequences were mapped against several previously published growth- and immune-related genes based on literature review (Chandhini & Rejish, 2019; Dam et al., 2020;Damzamnn et al., 2016; Guan & Qiu, 2020; Hu et al., 2016; Lin et al.,2019; Ma et al., 2016; Overturf et al., 2010; Palti, 2011; Vagner &Santigosa, 2011; Zhenzhen et al., 2014).Altogether, 82 growth- and 31 immune-related genes (113 genes in total) were downloaded from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/) and used as references.Subsequently, the genes were further filtered based on a stringent E-value cutoff of 10-10.
2.9. Ortholog inference and phylogenetic construction
TheTor tambroidesgenome was assembled in this study, while other genome sequences used were obtained from NCBI (National Center for Biotechnology Information) and summarized in Table 1.A total of 52 Actinopterygii species including one Sarcopterygii species as outgroup were included.
We had identified single-copy orthologous genes in each genome using BUSCO v5.3.0 (Benchmarking Universal Single-Copy Orthologs)(Manni et al., 2021) across all 52 Actinopterygii species and one Sarcopterygii species.BUSCO was run with orthologs in actinopterygii_odb10 (updated 2021-02-19) using default parameters.All BUSCOs found in both single-copy and multi-copy for each species wereused for phylogenetic analysis.Subsequently, BUSCO sequences were individually aligned with MUSCLE (Multiple Sequence Comparison by Log-Expectation) across all 53 species (Edgar, 2004).Gaps and unmatched sites were removed from the resulting multiple sequence alignments (MSAs) using trimAl (Capella-Guti′errez et al., 2009).These trimmed MSAs were concatenated into a supermatrix.A maximum-likelihood (ML) phylogenetic tree was inferred from the supermatrix using IQ-TREE (Nguyen et al., 2015).Molecular Evolutionary Genetic Analysis (MEGA) (Windows 10 system) v11.0.10 was used to analyse and view the generated phylogenetic tree (Kumar et al.,2018).
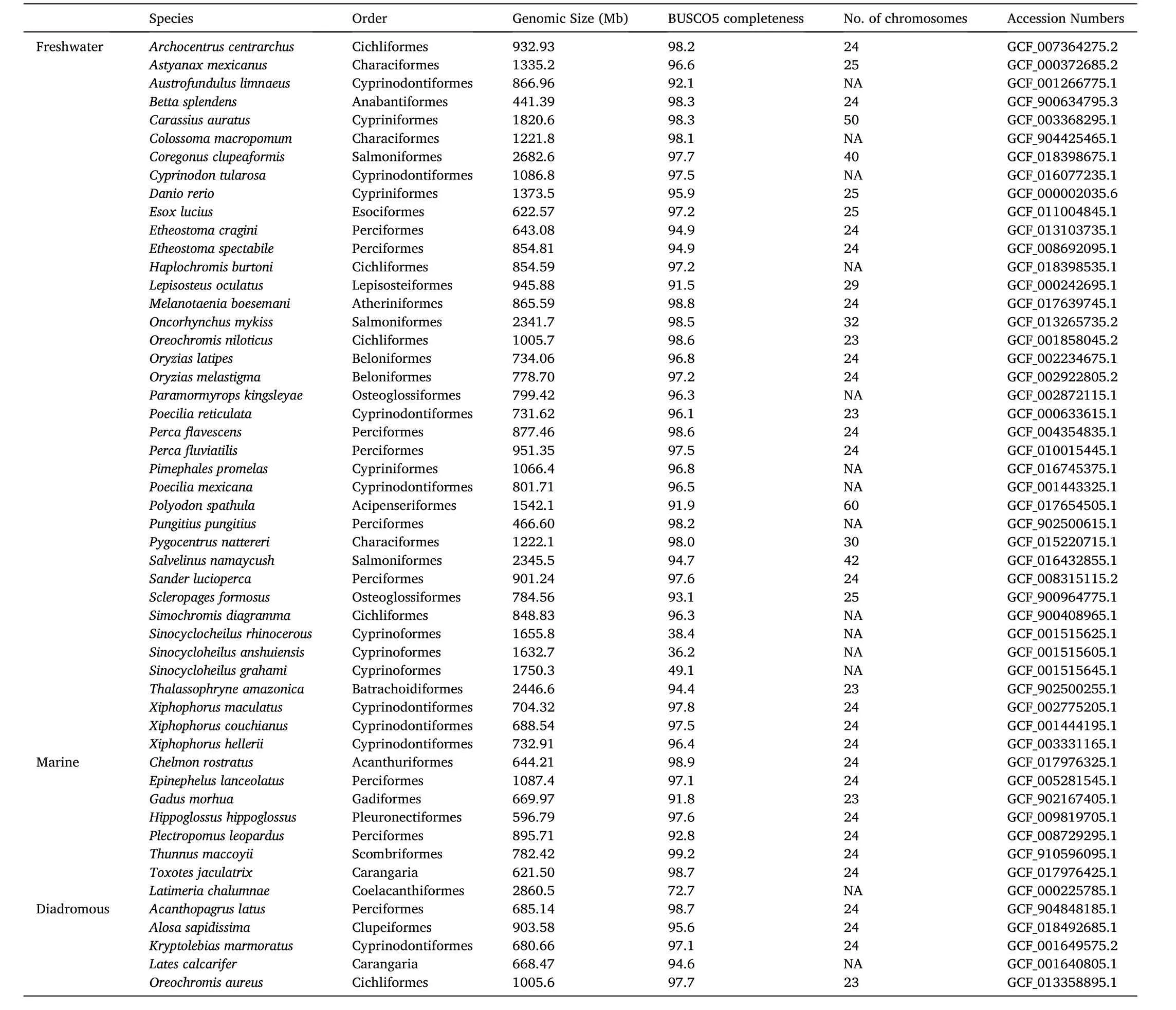
Table 1 Available genomes of fish species and their genome statistics used in the Maximum-likelihood (ML; IQ-TREE) tree construction.A total of 50 species with one Sarcopterygii species as outgroup were reported and classified into their respective residing environment, including freshwater, marine, or diadromous.
The whole genome sequence and raw sequencing reads ofT.tambroidesreported in this study are publicly available under the NCBI Bioproject PRJNA708136.The genome annotation and polished genome assembly generated in this study, were also uploaded in Zenodo database (https://doi.org/10.5281/zenodo.6371903).
3.Results & discussion
3.1. Characterisation of T. tambroides genome
The genomic sequencing and assembly statistics of the targetT.tambroidesin this study were summarized in Table 2.The total contig length is 1,235,011,685 bp with 44,726 contigs.The longest contig length recorded is 445,922 bp.The contig GC content documented in this study is 36.57%.
The assembled clean reads of theT.tambroidesgenome were subjected to K-mer analysis using Jellyfish software and visualised using GenomeScope (K-mer:31), as shown in Fig.1.There is no heterozygous peak generated at 23, and a low heterozygosity level of 0.194% was recorded.The depth of the homozygous peak can be observed at 46,which accounts for the identical 31-mers from both strands of DNA.The kmer-based statistical approach had revealed that 24.675% of the genomic content is repeated whereas 75.325% of the content is unique.The genome size ofT.tambroidescan be predicted by division of K-mer number over K-mer depth.The k-mer number detected in this study is 69 Gb.Therefore, the expected genome size is predicted as 1.23 Gb.The current assembled genome showed an Illumina mapping rate of 86.59%suggesting that a majority of the genome have been assembled.
It had been reported previously thatT.tambroideshas 2n = 100(Donsakul & Magtoon, 2008).In this study, the genome size ofT.tambroidesis reported as 1.23 Gb, which was found to be lower than otherCyprinidaetetraploid, such as common carp (Cyprinus carpio) at 1.83 Gb (Xu et al., 2014), goldfish (Carassius auratus) at 1.85 Gb (Chen et al., 2019).A tetraploid genome is more likely to retain a larger portion of duplicated genes due to whole-genome duplication event (Xu et al.,2014).Besides, its GC% content reported (36.55%) was slightly lower than the 37.4% and 37.3% seen inC.idellaandC.carpio.In general, a more outstanding GC content was found in seawater (usually above 40%) than freshwater fish (less than 40%), and also in migratory than a non-migratory species (Lu & Luo, 2020).Thus, it is suggested that the genomic GC content may be influenced by different living environments(Lu & Luo, 2020).Besides, the contrary relationship between genomicsize (1.23 Gb) and GC content (36.55%) was shown in genome ofT.tambroides.However, such assertion was insignificant based on a study reviewing 14 species, thus suggesting the collection of more genomic data for further validation (Lu & Luo, 2020).The gene number,gene length, exon length, coding region sequence (CDS) length and protein length ofTor tambroidesin comparison with those closely related fish species were shown in Fig.2.

Table 2 Genomic sequencing and assembly statistics.
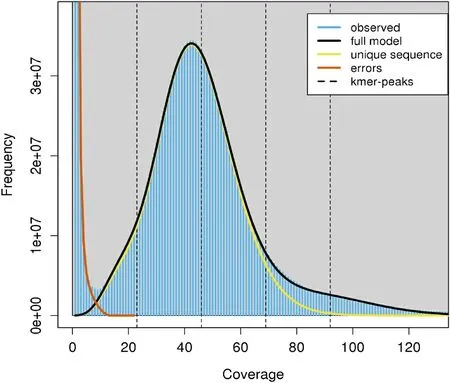
Fig.1.Estimation of genome size, repeat content and heterozygosity by GenomeScope, based on 31-mers (read length =150 bp; kmer max coverage at 1000).The y-axis had demonstrated the amount of K-mers found at each corresponding depth on the x-axis.
3.2. Repeat-content analysis
RepeatMasker and RepeatModeler were used to characterize the repetitive sequences within the genome ofT.tambroides(Table 3).Repetitive elements masked within the genome include satellites, simple repeats, sequences of low complexity, and transposable elements (TEs).The total interspersed repeats were recorded as 1,235,011,685 bp,consisting of 4,118,572 elements (39.84%).TEs are divided into two main classes, based on the presence (class I: retrotransposon) or absence(class II: DNA transposon) of RNA intermediate were found within the genome as well.Retrotransposons can be further classified into longterminal-repeat (LTR) retrotransposons.Depending on the type of reverse transcriptase (RT) possessed by each LTR retrotransposon, they can be categorized into Ty1/copia, Bel, and Ty3/gypsy groups.Another group is the tyrosine recombinase retrotransposon, having similar properties to those of LTR retrotransposons but possesses integrase instead of recombinase.The third classification is non-LTR retrotransposons with the absence of inverted or tandem terminal repeats.These have poly(A) tails at 3′end and variable deletions at 5′ends which encode open reading frame (ORF), which are prone to mutation (Eickbush & Jamburuthugoda, 2008).For instance, these elements include LINEs (long interspersed nucleotide elements) and SINEs (short interspersed nucleotide elements).LINEs are autonomous and about 5–10 kb in length and while SINEs are non-autonomous and about 100–400 bp in length (Tang, 2007).Penelope-like element (retroelement) were found as the lowest number of repeats observed, which accounted for 0.04% of the genome.As one of the retrotransposons, it was first identified fromDrosophila viriliswhich is responsible for the hybrid dysgenesis syndrome when it transposes and causes mutation (Evgen’ve et al., 1997).Both Penelope-like retrotransposon and LINEs are very diverse in structures as compared to other elements.They both own a Uri domain(GIY-YIG) and make use of the ends of chromosomes as primers in reverse transcription.
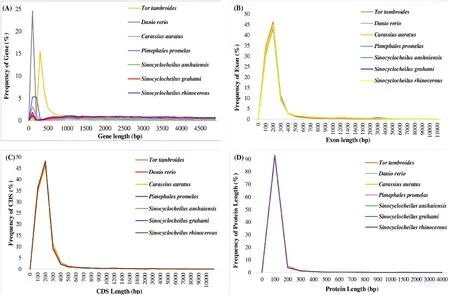
Fig.2.Length distribution comparison on total gene, exon, CDS and protein of annotated genes of Tor tambroides. Length distribution of gene (A), exon (B), CDS (C)and protein (D) was compared to Danio rerio, Carassius auratus, Pimephales promelas, Sinocyclocheilus anshuiensis, Sinocyclocheilus grahami and Sinocyclocheilus rhinocerous.
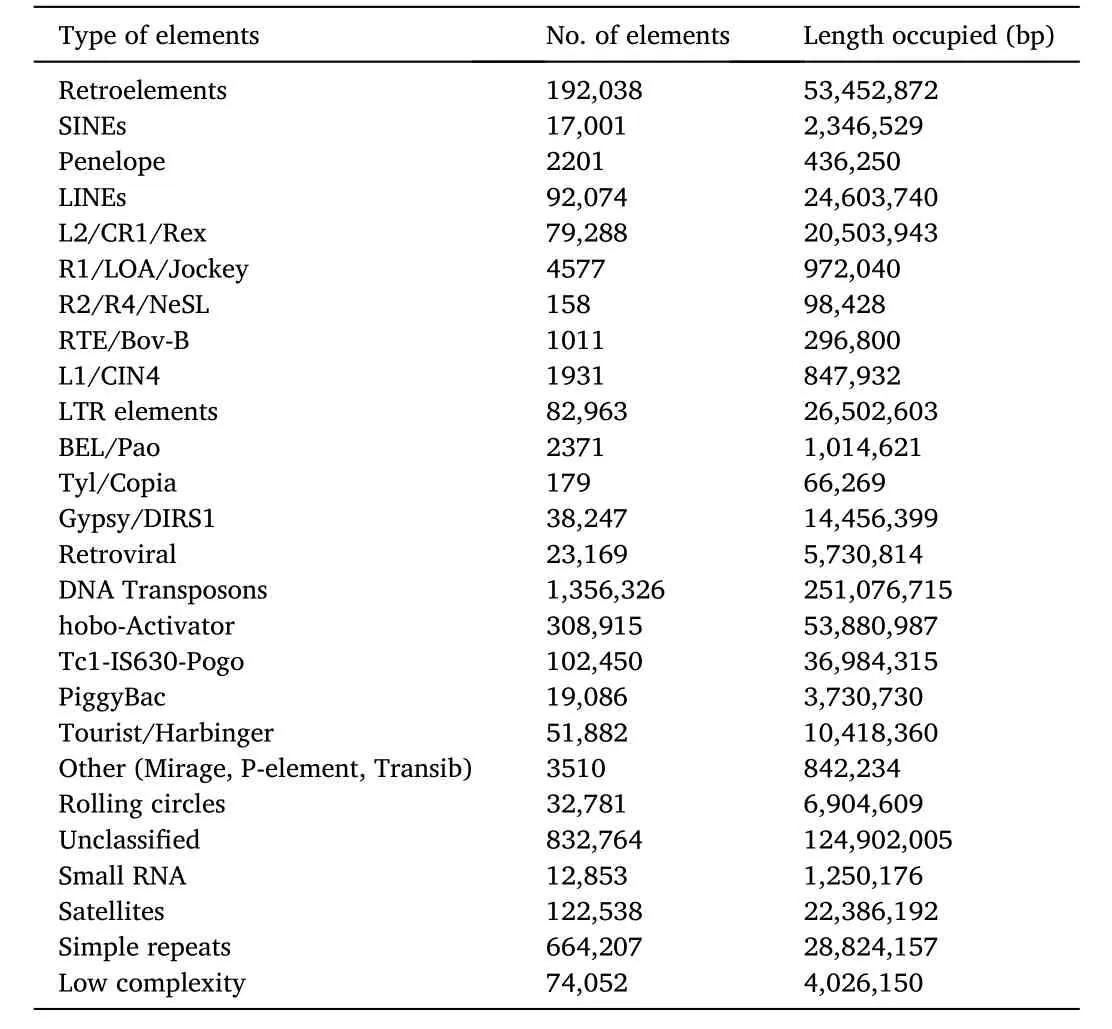
Table 3 Summary of repeats within the genome of T. tambroides.
The highest number of repeats were reported as DNA transposons(20.33%) followed by unclassified repeats (10.11%).DNA transposons have a short terminal inverted repeats with a long ORF encodes for transposase and DNA binding mechanisms.DNA transposon utilize ‘cutand-paste’ mode of action where the elements is cleaved and replicated then transported to another location by transposase, which were observed in most eukaryotes.Class II TEs were reported in a greater number than class I TEs inT.tambroideswhich is in consistent with the findings done by Yuan et al.(2018) stating that class II transposons were more abundant in freshwater than marine fish.Such species enrichment repetitive elements associated with living environment thus suggest the importance of these elements in genomic evolution and underlying potential in fish adaptation to their respective habitats (Yuan et al., 2018).Besides, it can also be said that the frequent stress including floods and droughts in freshwater ecosystem could accelerate transposition and further enhance host adaptation to the environment through generation of new genetic variants (Schrader et al., 2014).
However, rolling circle mechanism is used by other eukaryotic elements during DNA transposition as well and is similar to that occurring in prokaryotes (Kapitonov & Jurka, 2001).Both mechanisms were observed within the genome ofT.tambroidesbut rolling circles were in a lower occupied sequences as compared to DNA transposon.Another transposon element identified withinT.tambroidesgenome is PiggyBac transposon.Its transposase activity was observed inDrosophila melanogasterwhen the mutator elements present on the X chromosome in males were used through aHermes-based jump element withα-1-tubulinas promoter (Nyaku et al., 2021).Low-complexity repeats were observed in 0.33% within the genome ofT.tambroides.It was observed in many genomes and among protein families (Coletta et al., 2010) with low diversity in its encoded amino acid sequences with variations ranges from one or many amino acids at specific positions (Nyaku et al., 2013).Simple repeats accounts for 2.33% of theT.tambroidesgenome and could either be microsatellites or minisatellites (Nyaku et al., 2021).These simple repeats could contribute to the evolution within organisms(Vinces et al., 2009).
These repetitive elements accounted for 39.84% inT.tambroidesgenome, which is likely to be associated with its genome duplication(Yuan et al., 2018).It is found to be in accordance to the repetitive elements found across order Cypriniformes species reporting to be around 35%–40%, includingDanio rerio,Cyprinus carpio,Sinocycloheilus graham,Sinocycloheilus rhinocerous,Sinocycloheilus anshuiensisandPimephales promelas(Yuan et al., 2018).Within teleost, researchers had found out that the increase of repetitive elements may be the factor for the expansion of fish genome size as observed across 52 teleost species(Yuan et al., 2018).Having a high repetitive element content within the genome could speed up the generation of novel genes for adaptation purposes.However, an excessive amount of it would bring to abnormal combination and splicing, resulting in unstable genomes (Hong, 1998).In short, it is unfavourable for unlimited expansion of repetitive elements since it causes an increase in genomic size.It should be limited and shaped under specific natural selection (Yuan et al., 2018).
3.3. SSR analysis
SSRs allow faster adaptation to environmental stress through increment of DNA quantity and raw material for adaptative evolution during genome evolution.Thus, it can be said the mutation rate of microsatellite is dependent on the repeated unit length with a more common observation of mono- and dinucleotide repeats as compared to other repeats with longer lengths due to respective stability (Schl¨otterer,2000).The frequency of tetranucleotide was higher than trinucleotide within the genome ofT.tambroides,which was shown in other ray-finned fishes as well (Lei et al., 2021).Less occurrence of trinucleotide SSRs repeats can be due to its attribution as a triplet code to form part of the gene and also the presence of a mismatch repair system in the exonic region to maintain greater trinucleotide repeats (Lei et al., 2021).
SSR repeats with poly (A/T) tracts were found in a greater abundance than repeats with poly (G/C) tracts in other ray-finned fishes across all types of SSRs including dinucleotide, trinucleotide, tetranucleotide and pentanucleotide (Lei et al., 2021).The higher frequency of poly (A) can be due to the re-integration of the processed genes from mRNA back into the genome with an attached poly (A) tail, while poly (G/C) is not included in this integrative mechanism (Lei et al., 2021).In addition,greater poly (A) occurrence can be explained through the formation of pseudogenes and its necessity in the universal retrotransposon (Toth et al., 2000; Borodulina et al., 2016).The (GC) repeats are more stable than (AT) repeats thus increasing the difficulty to be slipped during replication (Gur-Arie et al., 2000).Thus, GC-rich sequences are ubiquitous among coding regions in both vertebrates and prokaryotes (Oliver& Marin, 1996).
Dinucleotides AT/TA is the common microsatellites repeat found across the fish genomes (Lei et al., 2021) which is observed within the genome ofT.tambroidesas well.Dinucleotide AC/GT was placed second in the total dinucleotides proportion.Such dinucleotide microsatellite distribution is in agreement with those found inDanio rerio,containingmore (AT) repeats followed by (AC) repeats (Chong et al., 2011).However, (AC) repeats are in a greater distribution than (AT) repeats inLates calcarifer,Oryzias latipesandDichotomyctere nigoviridis(Chong et al., 2011).This can be explained through the similar order thatD.rerioandT.tambroidesbelong to.Furthermore, for trinucleotide repeats, the occurrence of (CCG) n (16 counts) and (ACG) n (2572 counts; 5%) repeats were rare inT.tambroidesas well.This phenomenon can be explained by the presence of the highly mutable CpG dinucleotide within the motif due to methylation (Lei et al., 2021).(AAT) n repeats(17260 counts; 28.78%) recorded as the greatest number of trinucleotide repeats as it has greater hairpin propensities (Chong et al., 2011).Such nucleotide sequences are self-complementary which they can base pair to form hairpins or loops and stabilize strand slippage (Moore et al.,1999).
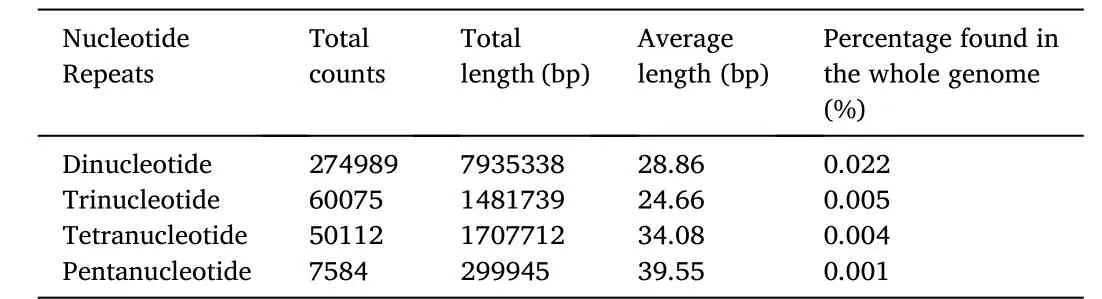
Table 4 The number, total length and average length of five different types of SSRs found within the genome of T. tambroides.
While in tetranucleotide repeats, the G+C content of SSRs was observed in a lower frequency because of its influence on the mutation rate as there is no statistical significance between 25% G+C content but each was significant difference from the 50% G+C repeat content(Eckert et al., 2002).
The repetitive element found within the genome ofT.tambroideswas reported as 4.15% in this study.It had been reported previously that the expansion of repetitive elements would cause a further expansion in the fish genome Therefore, it can be said that the fish genome size is positively correlated with the repetitive elements (Yuan et al., 2018).However, a study reviewing the SSR across 14 fish species contradicts the statement (Lei et al., 2021).The generation of novel genes for adaptation can be accelerated with the presence of high repetitive element content, for instance in salmon, which is likely to be associated with genome duplication (Lien et al., 2016).Besides, the variation observed within microsatellites can be due to differential selective constraints causing accumulated preference for different microsatellite types (Lei et al., 2021).However, overburden could cause abnormal recombination and splicing, resulting in genome instability (Yuan et al.,2018).In short, it can be concluded that the repetitive elements must be limited to shape under specific natural selection by the environment.Its unambiguous role in genomic function still remains to be explored.
3.4. Functional annotation of T. tambroides
For functional annotation ofT.tambroidesgenome, coding region was extracted using Interproscan (Jones et al., 2014), generating raw amount of 76,419 predicted coding sequences.Subsequently, the predicted protein sequences were annotated using eggNOG mapper to map against GO, KEGG, and COG databases.Table 5 shows the number of predicted protein sequences to either GO, KEGG or COG.Altogether, 20,341 predicted protein sequences annotated to at least one of the databases and all the databases.A total of 39,902 (74.48%), 24,040(44.87%), and 53,511 (99.88%) of predicted protein sequences were annotated to GO, KEGG, and COG databases respectively.Out of a total of 53,573 annotated predicted protein sequences, 44,519 (83.10%) of the them had been found to have a significant match to at least one of the databases while 20,342 (37.97%) predicted protein sequences portrayed a notable match to all the three databases.Fig.4 illustrates the distribution of predicted protein sequences across GO, KEGG, and COG databases.
So nothing was left for them but to take their departure to the cottage, which stood in the midst of a dark forest, and seemed to be the most dismal8 place upon the face of the earth
Annotation ofT.tambroidesgenome to each main ontology of GO database was shown in Fig.S1, including biological process, molecular functions, and cellular components.Under biological process, metabolism (4460; 10.74%) had the greatest count, followed by development(3335; 8.03) and catalytic activity (2141; 5.15%).On the other hand, a total of 1239 counts (2.98%) were responsible for binding, 757 (1.82%)accounted for transferase activity and 690 (1.66%) accounted for protein binding, under the molecular function category.Furthermore,under the cellular components category, 2130 (5.13%) were accounted for cell organization and biogenesis while 1659 (3.99%) and 1274(3.07%) were categorized as cell and intracellular respectively.
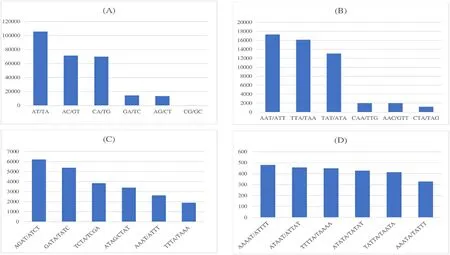
Fig.3.SSRs percentage graph with selected top six frequency SSRs from each group: (A) dinucleotide repeats, (B) trinucleotide repeats, (C) tetranucleotide repeats,(D) pentatetranucleotide repeats.
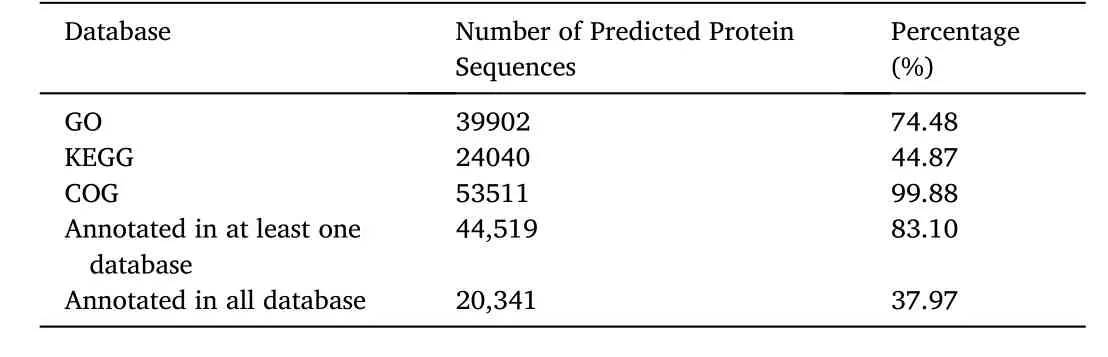
Table 5 Functional annotation of predicted protein sequences to the various database.
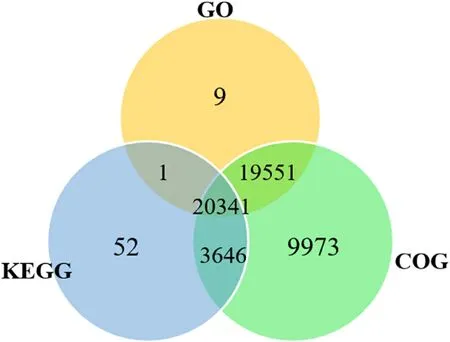
Fig.4.Venn diagram showing differences and similarity of predicted protein sequences of T. tambroides annotated to GO, KEGG, and COG databases.
Another annotation was performed towards a widely-used reference database KEGG equipped with multiple pathways for better integration and interpretation of large-scale datasets.T.tambroidesgenome had successfully mapped towards 304 known KEGG pathways (Fig.S2),including organismal system, cellular processes, environmental information processing, genetic information processing and metabolism.Out of the five main aforementioned categories, the largest count (35694;38.36%) is from organismal system whilst genetic information processing (4157; 4.55%) had the lowest count.The categories reported on the greatest number of counts were signal transduction from environmental information processing (17978; 19.67%), endocrine system from organismal system (9035; 9.89%) and immune system (7960; 8.65%)from organismal system.Fig.S3 depicts the top ten KEGG cluster components found in each main aforementioned category.The top three largest count can be observed in metabolic pathway (4126; 4.51%),microbial metabolism in diverse environment (1510; 1.65%) and PI3KAkt signaling pathway (1406; 1.54%) from signal transduction.
Fig.S4 illustrates the classification of 76,419 predicted protein sequences towards COG database consisting of clusters of orthologous groups.There were altogether 25 COG classifications and can be grouped under four main clusters: information storage and processing(8971; 15.13%), cellular processes and signaling (24680; 43.08%),metabolism (8023; 14.00%) and poorly characterized (15920; 27.59%).Overall, the genome ofT.tambroidesis enriched with gene families in the categories of signal transduction, endocrine system, immune system and metabolic pathways.It is found to be consistent with the common carp genome (Xu et al., 2014) and Javan mahseer transcriptome (Lau et al.,2021b).
3.5. Immunity-related annotation
To characterize a comprehensive defense landscape ofT.tambroidesagainst pathogenic infections, the genomic sequences ofT.tambroideshad been functionally annotated to identify pathways and genes associated with the fish immunity.GO classification had revealed on 76,419 predicted protein sequences were functionally annotated to immunerelated GO terms.Among the annotated categories, regulation of immune system (3926), immune response (3608) and immune system development (2415) had reported on the greatest counts.KEGG pathway analysis had revealed 41 immune-related pathways, including MAPK signaling pathway, Toll-like receptor (TLR) signaling pathway,Wnt signaling pathway, NOD-like receptor signaling pathway and so on(Fig.5) (Zhenzhen et al., 2014).
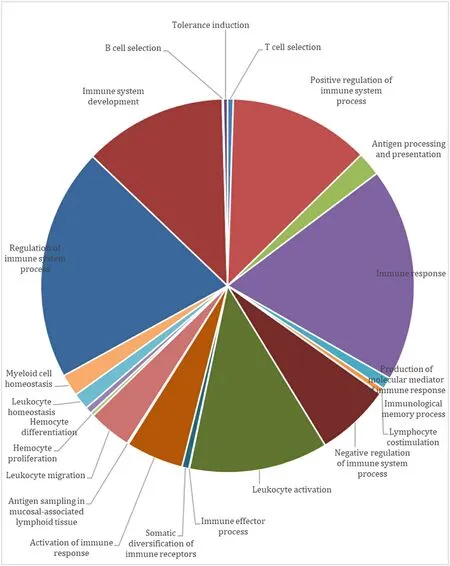
Fig.5.Annotation of T. tambroides genome to known immune-pathway within GO databases.
Among all the components in the fish immune system, TLR family is an essential type of pattern-recognition receptors expressing on antigenpresenting cells, involved in innate immune response and the subsequent promotion of adaptive immunity (Akira et al., 2006).We had successfully identified 11 different interleukins (IFNs) and 14 TLR genes matched to the genome ofT.tambroides(Table S1).To date, there were at least 21 types of TLRs (TLR1-5, 5S, TLR7-9, TLR13, TLR14, TLR18-23 and TLR25-28) identified within fishes including both orthologs of mammalian TLRs and species-specific TLRs (Nie et al., 2018).Vertebrate TLRs can be classified into six major subfamilies, including subfamily TLR1, TLR3, TLR4, TLR5, TLR7 and TLR11 (Roach et al., 2005).Within TLR1 subfamily, TLR1 and TLR2 had been identified in the genome ofT.tambroides.In mammals, TLR1 is responsible in the recognition of triacylated lipoproteins and mycobacterial products by binding to TLR2 to form a heterodimer (Randelli et al., 2008).Both TLRs had been characterized previously in other fishes as well including zebrafish(Danio rerio) (Jault et al., 2004), common carp (Cyprinus carpio) (Fink et al., 2016) and grass carp (Ctenopharyngodon idella) (He et al., 2016).TLR2 signaling pathway is shown to involve in the recognition of probioticsPsychrobactersp.And activation of mucosal immune system in orange-spotted grouper (Nie et al., 2018).TLR14 is found to be associated with immune response against gram negative and positive bacterial infection as well as viral infection (Hwang et al., 2011).
Next, TLR3 is responsible for innate immune response that detect double-stranded RNA (dsRNA), endogenous cellular mRNA and sequence-independent small inferring RNAs (Sahoo et al., 2012).As the dsRNA binds to TLR3, production of interferon and inflammatory cytokine will be induced in fish cells (Matsuo et al., 2008).In addition,the members of subfamily TLR4 are mainly associated with lipopolysaccharide (LPS) recognition and are known as the best characterized pattern recognition receptor (PRR) (Lu et al., 2008).A significant upregulation of TLR4 was observed within the muscles and liver of adult grass carp after grass carp reovirus (GCRV) infection, indicating its role in immune function (Huang et al., 2012).As one of the members of familyCyprinidae,T.tambroideswas detected with its TLR4 which partially further support the speculation of TLR4-LPS signaling pathway appeared after the divergence of fish and tetrapod (Nie et al., 2018)when sequencing of pufferfishes (bothTakifugu rubripesandTakifugu niroviridis) and stickleback (Gastrosteus aculeatus) show no sign of TLR4(Li et al., 2012; Oshiumi et al., 2003).TLR5 subfamily consisted of only one member, TLR5, presented in both membrane-bound form (TLR5M)and non-transmembrane soluble form (TLR5S) (Tsujita et al., 2004).In teleost, TLR5M is highly expressed in head kidney, spleen, liver and brain tissues while TLR5S is detected mainly in liver (Bai et al., 2017).TLR5 recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) and further activate MyD88-dependent TLR signaling pathway during heat and cold shock,which had been characterized in Indian major carp (Basu et al., 2015).
The TLR7 subfamily comprises of TLR7, TLR8 and TLR9, which are structurally similar to mammalian homologs.There is a reduced in the expression of TLR7 in blood and lymphoid tissues of zebrafish with an elevated expression in TLR8 after 8-week infection with pathogenMycobacterium marinum(Meijer et al., 2004).Furthermore, upregulated TLR9 expression can be observed in spleen and kidney tissues followingVibrio parahaemolyticusinfection in common carp (Kongchum et al.,2011), large yellow croaker (Yao et al., 2008) and gilthead sea bream(Franch et al., 2006).Subfamily TLR11 is consisted of numerous ‘fish--specific’ TLRs.For instance, upregulation of TLR22 is seen in a variety of common carp tissues when it is challenged withAeromonas hydrophila,further indicated the essential role of TLR22 in systemic and mucosal defense after viral or bacterial infection (Wang et al., 2017).
The major histocompatibility complex (MHC) are the important molecules for the recognition of foreign substances via binding peptide fragments from pathogens and presenting them for T cell elimination(Neefjes et al., 2011).Both MHC class I and II had been identified in the genomic dataset as well.MHC genes serve as a candidate disease resistance marker due to their highly polymorphic characteristics in teleost(Langefors et al., 2001).Both TLR and MHC were also found within the transcriptomic dataset ofTrachinotus ovatus(Zhenzhen et al., 2014).Thus, it is believed that further analysis of both genes will be able to provide insights into the immune system ofT.tambroidesas well as other teleosts.
3.6. Growth-related annotation
To tap into the growth-related aspect of theT.tambroidesgenomic landscape, we functionally annotated the genome based on previously characterized growth-related genes.Table S2 shows the mapped BLAST results of immune-related genes on the genome ofT.tambroideswhere there were 82 genes that had passed the E-value cutoff filter of 10-10.KEGG pathway analysis revealed 19 pathways associated with growth,including pathways in cancer, insulin signaling pathway, endocytosis,focal adhesion, and mTOR signaling pathway (Zhenzhen et al., 2014)(Fig.6).
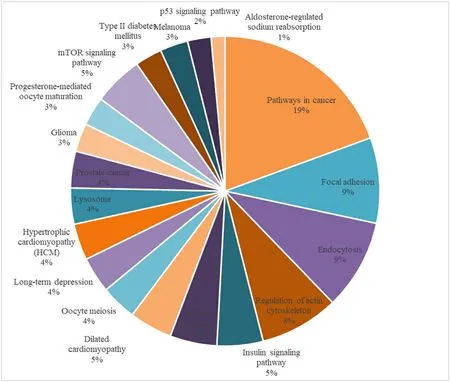
Fig.6.Annotation of T. tambroides genome to known top 20 growth-related pathway in KEGG database.
Thus, it can be said that a significant similarity had been exhibited with previously published immune-related genes.For instance, growth hormone (GH) and insulin-like growth factor-binding protein (IGFBP)which are responsible for the regulation of GH and IGF (Zhenzhen et al.,2014) were detected in high similarity in the genome ofT.tambroides(Table S2).Somatostatins were found to have an inhibitory role in promoting the release of GH (Li & Lin, 2010).
It was found out that regulation of appetite, protein and lipid metabolism, weight gain and muscle growth are also part of the complex growth process (Zhenzhen et al., 2014).A few genes related to appetite and muscle were detected in the genome ofT.tambroides,for instance,neuropeptide Y (NPY), pro-opiomelanocortin (POMC), leptin and myostatin (Table S2).These genes play various roles whereNPYandPOMCare exerting antagonistic roles in stimulating or inhibiting feeding(Zhenzhen et al., 2014).In addition, myostatin is a negative regulator of muscle growth and its polymorphism is associated with growth traits(Nadjar-Boger & Funkenstein, 2011).In addition, leptin is responsible for the regulation of energy intake and usage (Zhenzhen et al., 2014).
Moroever, genes that encode for proteolytic digestive enzymes(chymotrypsin-like elastase,cela) and are related to protein metabolism(cathepsin L,clsLand cathepsin K,clsK) were detected in the genome ofT.tambroidesas well.In addition, a number of lipid metabolism regulation genes were found in the genome ofT.tambroides,including lipase C (lipC), phospholipase A2 (pla2), elongation of very long chain fatty acid family 6 (elovl6), apolipoprotein B (apob), acetyl-CoA carboxylase(ACACA,ACACB) and fatty acid synthase (FASN).This can be due the fact thatT.tambroidesis a semi-fatty fish and it contains 4.6–5.2% of muscle crude lipid (¨Ozogul & ¨Ozogul, 2007), indicating the importance of lipid metabolism in this fish.Lipase is a key enzyme involved in lipid hydrolysis while apolipoprotein is a lipid-associated protein that regulates lipid homeostasis through the transport of triacylglycerol and phospholipid from the liver to other tissues (Infante & Cahu, 2007).ACACBwas found not only to be associated with fat yield and percentage, but it also plays role in protein yield as well (Han et al., 2018).FASNis an important element in lipid metabolism and its expression could vary due to fatty acid content in both fat and meat (Renaville et al.,2018).Furthermore, glucose-6-phosphatase (g6pc) which regulates carbohydrate metabolism were detected inT.tambroidesas well.These growth-related genes may serve as the possible molecular growth-related markers for further marker-assisted breeding.Further studies are required to confirm the roles of these genes in the growth of
T.tambroides.
3.7. Orthologs and phylogenetic inferences
The phylogenetic relationship ofT.tambroideswith other ray-finned fishes was inferred through the BUSCO supermatrix approach through single-copy orthologs.The BUSCO completeness of each species was summarized in Table S3.MUSCLE was used to align all the genomic sequences of 52 Actinopterygii species and one Sarcopterygii species as outgroup (Edgar, 2004).The phylogenetic tree was plotted using the maximum-likelihood model (ML; IQ-TREE) based on the single-copy and multi-copy orthologs (Fig.7).All the species except outgroup species,fall under class Actinopterygii but belong to 18 different orders,including Cypriniformes, Perciformes, Clupeiformes, Cichliformes,Characiformes, Cyprinodontiformes, Anabantiformes, Salmoniformes,Esociformes, Gadiformes, Pleuuronectiformes, Lepisosteiformes, Atheriniformes, Beloniformes, Osteoglossiformes, Batrachoidiformes with Coelacanthiformes and Acipenseriformes rooted as outgroup.As a member of the familyCyprinidae,T.tambroidesformed a monophyletic cluster with the species within order Cypriniformes, namely species from genusSinocyclocheilus,Carassius auratus,Pimephales promelasandDanio rerio.
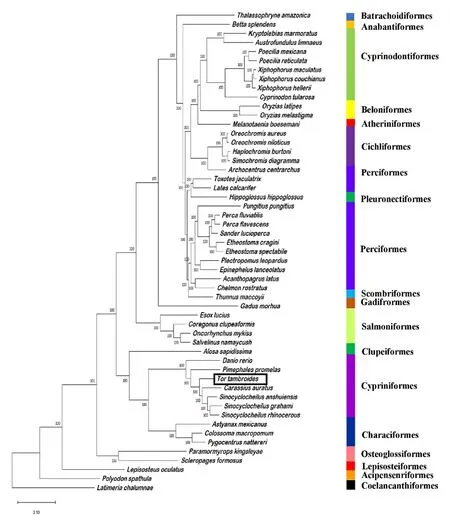
Fig.7.Maximum-likelihood tree plotted on 52 Actinopterygii species with one Sarcopterygii species as outgroup inferred from a supermatrix of 3640 BUSCOs.The species were displayed as order.
4.Conclusion
We present the first Malaysian mahseer (T.tambroides) genome assembled with low-coverage Nanopore long reads and high-coverage Illumina short reads.De novogenomic assembly had generated a draft genome with an estimated genome size of 1.23 Gb [87.6% BUSCO completeness (Actinopterygii_odb10)].Altogether, 392,760 SSRs had been identified from the genome with dinucleotide repeats AT/TA as the most common SSR.A total of 76,419 non-redundant coding sequences was predicted and used for later functional annotation.Predicted protein sequences had mapped to 304 known KEGG pathways with signal transduction as the highest representation.Furthermore, genes showing significant similarity to published growth- and immune-related genes were identified, hoping to serve as a potential marker for future molecular breeding ofT.tambroides.In addition, the first genome-based evolutionary relationship ofT.tambroidesbetween other ray-finned fishes had been inferred using a Maximum-likelihood tree.It is hoped that this genomic data ofT.tambroidescould be a more powerful tool that continues to resolve questions of species identification, evolutionary biology, morphological variations, sequences related to sex differentiation, growth, reproduction, and immune, which are helpful for further conservation ofTorspecies.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships which have or could be perceived to have influenced the work reported in this article.
CRediT authorship contribution statement
Melinda Mei Lin Lau:Writing – original draft, Data curation,Conceptualization.Leonard Whye Kit Lim:Data curation, Writing –original draft, Conceptualization.Hung Hui Chung:Conceptualization,Funding acquisition, Writing – review & editing.Han Ming Gan:Methodology, Conceptualization, Writing – review & editing.
Acknowledgements
This work was fully funded by Sarawak Research and Development Council through the Research Initiation Grant Scheme with grant number RDCRG/RIF/2019/13 awarded to H.H.Chung.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.aaf.2022.05.002.
 Aquaculture and Fisheries2023年5期
Aquaculture and Fisheries2023年5期
- Aquaculture and Fisheries的其它文章
- The effectiveness of light emitting diode (LED) lamps in the offshore purse seine fishery in Vietnam
- Effects of dietary phosphorus level on growth, body composition, liver histology and lipid metabolism of spotted seabass (Lateolabrax maculatus)reared in freshwater
- Immunomolecular response of CD4+, CD8+, TNF-α and IFN-γ in Myxobolus-infected koi (Cyprinus carpio) treated with probiotics
- Effects of transport stress on immune response, physiological state, and WSSV concentration in the red swamp crayfish Procambarus clarkii
- Expression of gastrin and cholecystokinin B receptor in Lateolabrax maculatus
- The competitiveness of China’s seaweed products in the international market from 2002 to 2017