
密度泛函活性理論預(yù)測單取代和多取代苯甲酸的pKa值
2011-12-08 06:02:46鐘愛國劉述斌
湖南師范大學(xué)自然科學(xué)學(xué)報 2011年1期
黃 鶯,鐘愛國,劉述斌
(1.湖南中醫(yī)藥大學(xué)藥學(xué)院,中國長沙 410208;2.臺州學(xué)院化學(xué)系,中國臨海 317000; 3.北卡羅萊納大學(xué)超算中心,北卡羅萊納州教堂山市,27599-3420,美國; 4.湖南師范大學(xué)化學(xué)化工學(xué)院,中國長沙 410081)
密度泛函活性理論預(yù)測單取代和多取代苯甲酸的pKa值
黃 鶯1,鐘愛國2,劉述斌3,4*
(1.湖南中醫(yī)藥大學(xué)藥學(xué)院,中國長沙 410208;2.臺州學(xué)院化學(xué)系,中國臨海 317000; 3.北卡羅萊納大學(xué)超算中心,北卡羅萊納州教堂山市,27599-3420,美國; 4.湖南師范大學(xué)化學(xué)化工學(xué)院,中國長沙 410081)
酸堿性是分子非常重要的物理化學(xué)性質(zhì)之一通常用pKa值來表示,但由于受穩(wěn)定性等諸多因素的制約從實驗上準(zhǔn)確測定一些分子的pKa值仍有困難.從理論和計算上尋找有效和可靠的預(yù)測酸堿性的方法仍然是目前文獻(xiàn)上活躍的課題.最近,我們從密度泛函活性理論的角度提出了一個簡單而有效的方法來計算分子的酸堿性.本文試圖把該方法應(yīng)用于苯甲酸體系,預(yù)測單取代和多取代苯甲酸的pKa值.我們發(fā)現(xiàn),本文的預(yù)測結(jié)果比文獻(xiàn)報道最好的計算值都要精確.
酸堿性;密度泛函活性理論;pKa值
The knowledge of pKavalues,the acid-base dissociation constant,as a measure of the strength of an acid or a base,is essential for the understanding and quantitative treatment of acid-base processes in solution.This infor ma-tion is relevant to chemical synthesis,pharmacokinetics,drug design,drugmetabolis m,toxicology,and environmental protection.To develop efficient and reliable computational models to predict pKavalues withab initioand density functional theory(DFT)approaches is still a daunting task,as an ongoing effort in the literature.[1-3]Very recently, we developed an effective method,both efficient and reliable,within the framework of density functional reactivity theory(DFRT)for the purpose.[4-5]In thiswork,we apply the approach to substituted benzoic acids,demonstrating its effectiveness and reliability in quantitatively forecasting pKadata for sometimes experimentally inaccessible systems.
In theory,the pKavalue of an acid,HA,is defined as the negative logarithm,pKa=-log10Ka,of the equilibrium constantKaof the acid dissociation reaction,HA?H++A-,withKa=[H+][A-]/[HA].In ther modynamics,the equilibrium constantKais related to the standard Gibbs free energy changeΔG°for this reaction,ΔG°= 2.303RTpKa,whereRis the gas constant andTis the temperature in Kelvin.In practice,the pKavalue can be computed through a ther modynamic cycle usingab initioand DFT methods,which is often computationally demanding.[1-3]The new approach we proposed was based on DFRT.[6-9]When a proton is dissociated from an acid molecule,the energy changeΔEof the entire system associated with the process can be expressed as follows[5]

whereρ(r)is the electron density andΔv(r)is the change of the external potential due to the dissociation of the proton,which can explicitly be expressed as

whereRHis the coordinate of the leaving proton and{Zi,Ri }are the nuclear charge and coordinates of the other nuclei in the acid molecule,respectively.Put together,this results in[5]

The right hand side of Eq.(3)is nothing but the molecular electrostatic potential(MEP)on the leaving proton nucleus,suggesting that theMEP of the leaving proton can serve as a linearpredictor of the pKavalue of the acid.Additionally,we demonstrated earlier that theMEP of the acidic atom,O1 in the study(Scheme 1),is also a reliable indicator of the acid's pKavalue[4].Also,a strong linear relationship between the acidic atom'sMEP and its valence natural atomic orbital(NAO)has been revealed[4].

Table 1 Reactivity descriptors from density functional reactivity theory employed to predict the pKavalues of 13 substituted benzoic acids and comparison of exper imental and predicted pKadata from thiswork and from the literature

Scheme 1Atomic numbering of substituted benzoic acids.R2-R6groups are hydrogen atoms by default.A substituted benzoic acid is denoted by its substituting group.For example,3-Br stands for the systemswith all R being H except for R3=Br.
In thisLetter,we applied the approach to predict the pKavalues for a series of substituted benzoic acid compounds(Scheme 1).We first consider the 13 singly substituted benzoic acids whose exper imental pKadata are known.[10]Computational details employed for these compounds are available elsewhere[4-5].Table 1 summarizes the results for these compounds(see Scheme 1 for their notation).Figure 1 displays the strong linear correlations betweenMEP and NAO for both O1(correlation coefficientR2=0.999 8)and H1(R2=0.999 8).Also shown in the Fig 1(c)is the least square fit result between MEP on O1 and the exper imental pKadata for the 13 species. W ithMEP and NAO descriptors from DFRT for both the acidic atom O1 and the leaving proton H1,we obtain two quantitative models from Table 1.The first fitted model,called“Model1”,takes the following for m,pKa=-38.916 MEPO1-863.91 withR2=0.937.W ith the data from H1,one can build a similar model,termed as“Model2”, pKa=-39.572 MEPH1-33.275 withR2=0.934.The standard deviation for both of our fitted models is 0.08,as shown in the last row of Table 1.Also,shown in the Table is the best theoreticalprediction result from the literature, which gives 0.66 as the overall standard deviation for this data set.The latter is much more computationally involved.It becomes apparent from these results thatour approach ismuch better in accuracy and much more efficient in computation.
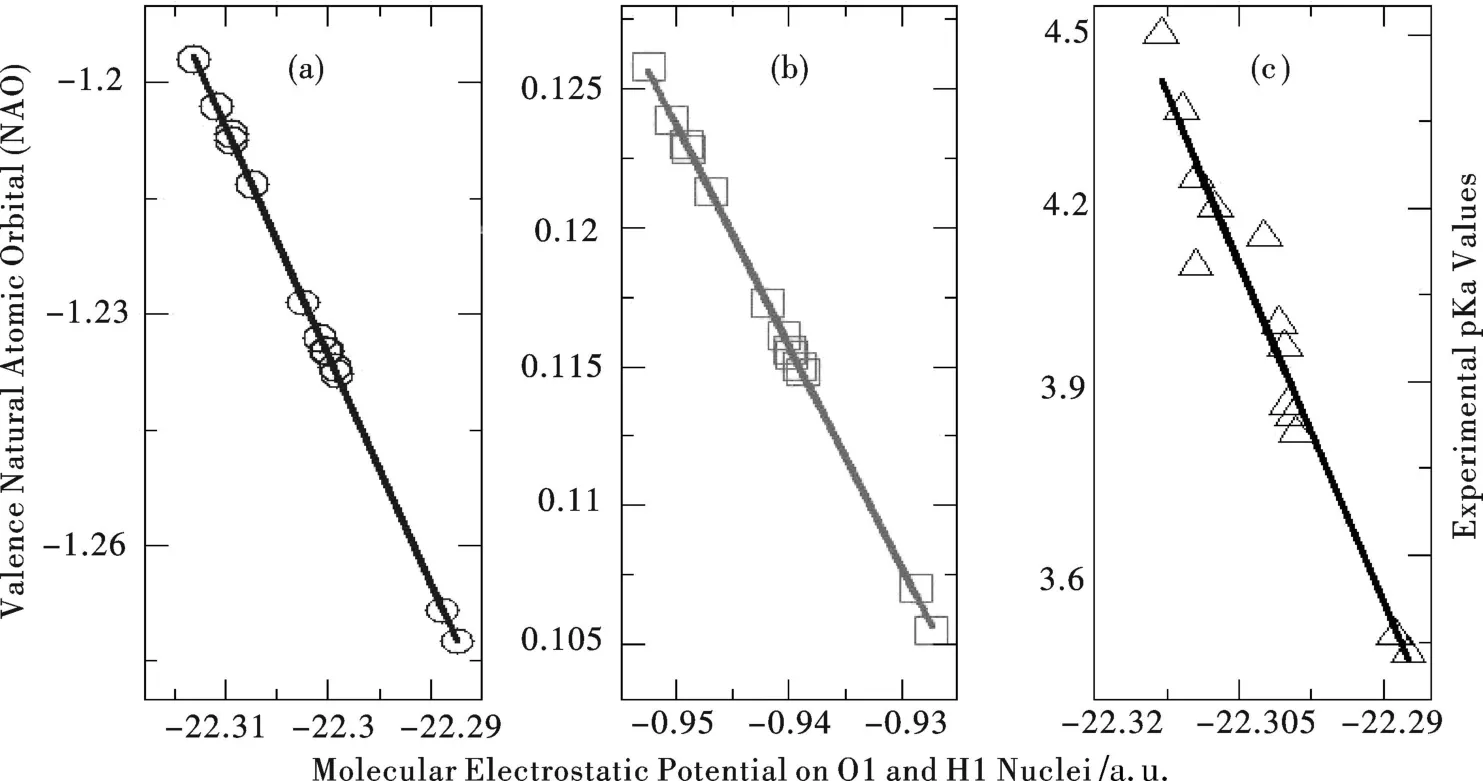
Figure 1 Strong linear correlations(a)betweenMEP and NAO ofO1;(b)be tweenMEP and NAO of H1; and(c)betweenMEP on O1 and exper imental pKadata for the 13 species in Table 1
Now,we use these two models above to predict the pKavalues for 20 singly,doubly and triply substituted benzoic acids,some ofwhich are experimentally inaccessible.The reason thatwe can do thismodel extrapolation is because these species belong to the same categoryofmolecules,so themodels can be employed to extrapolate the fitted data.Indeed,we found that the strong linear correlations be tween MEP and NAO of both O1 and H1 are still in place(not shown),with the correlation coefficientR2equal to 0.995 and 0.970,respectively,for the total of 33 data points.The predicted pKavalues from the two models are tabulated in Table 2,togetherwith theMEP and NAO descriptors from DFRT.It is seen from the Table that the more NO2groups are attached to the benzene ring,the low-er the pKavalue,and that the moreMe and OMe groups are substituted to the benzene,the higher the pKavalues. The smallest pKavalue was found for the 2,3,4-NO2compound(pKa=2.44 and 2.41,respectively from the two models)and the largest pKavalue was obtained for 2,4-OMe with both models giving pKa=4.88.These observations are consistentwith the conventional chemical intuition that electron donating groups such as-CH3and-OCH3increase the benzoic acid's pKavalue,whereas electron accepting groups such as-NO2and-F decrease its pKavalue. These results validate the effectiveness and reliability of our new approach to quantitatively predictmolecular acidity.

Table 2 Reactivity descriptors from density functional reactivity theory employed to predict the pKavalues of 10 substituted benzoic acidswhose experimental pKadata are not available
Acknowledgements
Thiswork was supported in part by a special funding from Hunan Normal University to Xiaoxiang Scholars. Helpful discussion with Robert G.Parr ofUniversity ofNorth Carolina at Chapel Hill is gratefully acknowledged.We thank the Editor of this Journal for the kind invitation.
[1] JORGENSEN W L,BR IGGSJM,GAO J.A priori calculationsofpKa's fororganic compounds inwater.The pKaof ethane[J]. J Am Chem Soc,1987,109:6857-6858.
[2] L IM C,BASHFORD D,KARPLUSM.Absolute pKacalculationswith continuum dielectricmethods[J].J PhysChem,1991,95: 5610-5620.
[3] POTTER M J,G ILSON M K,MCCAMMON J A.Smallmolecule pKaprediction with continuum electrostatics calculations[J].J Am Chem Soc,1994,116:10298-10299.
[4] L IU SB,PEDERSEN L G.Estimation ofmolecular acidity via electrostatic potential at the nucleus and valence natural atomic orbitals[J].J Phys Chem A,2009,113:3648-3655.
[5] L IU S B,SCHAUER C K,PEDERSEN L G.Molecular acidity:A quantitative conceptual density functional theory description [J].J Chem Phys,2009,131:164107.
[6] PARR R G,YANGW.Density-functional theory of atoms and molecules[M].New York:Oxford University Press,1989.
[7] GEERL INGS P,DE PROFT F,LANGENAEKER W.Conceptual density functional theory[J].Chem Rev,2003,103:1793-1874.
[8] CHATTARAJ P K,SARKAR U,ROYD R.Electrophilicity index[J].Chem Rev,2006,106:2065-2091.
[9] L IU SB.Conceptual density functional theory and some recent developments[J].Acta Phys Chim Sin,2009,25:590-600.
[10] AL I S T,KARAMAT S,KONA J,et al.Theoreticalprediction of pKavaluesof seleninic,selenenic,sulfinic,and carboxylic acids by quantum-chemicalmethods[J].J Phys Chem A,2010,114:12470-12478.
[11] HOLL INGS WORTH C A,SEYBOLD P G,HADAD CM.Substituent effects on the electronic structure and pKaof benzoic acid [J].Int J Quantum Chem,2002,90:1396-1403.
Predict ing pKaValues for S ingly andMultiply Substituted Benzoic Acids with Density Functional Reactivity Theory
HUANG Ying1,ZHONG Ai-guo2,L IU Shu-bin3,4*①
(1.School of Phar macy,Hunan University of ChineseMedicine,Changsha 410208,China;
2.Depar tment of Chemistry,Taizhou College,Linhai 317000,China;
3.Research Computing Center,University ofNorth Carolina,Chapel Hill,North Carolina 27599-3420,USA;
4.College of Chemistry and Chemical Engineering,Hunan NormalUniversity,Changsha 410081,China)
Molecular acidity is one of the most important physicochemical properties of molecules, yet difficult to exper imentally measure and computationally predict in many cases.To develop an efficient and reliable approach from theory to calculate molecular pKavalues is challenging and still of recent interest in the literature.A novel approach from density functional reactivity theory has recently been developed by us.In thisLetter,we apply the approach to substituted benzoic acids.Our results show that the new approach is able to provide quantitative predictions for their pKavalues,better than the best computational results reported in the literature.
molecular acidity;density functional reactivity theory;pKavalue
O641
A
1000-2537(2011)01-0052-04
2010-12-28
*通訊作者,E-mail:shubin@email.unc.edu
(編輯 楊春明)
猜你喜歡
大眾文藝(2021年8期)2021-05-27 14:05:54
云南化工(2020年11期)2021-01-14 00:50:52
大眾文藝(2020年11期)2020-06-28 11:26:50
大眾文藝(2019年16期)2019-08-24 07:54:00
中學(xué)化學(xué)(2019年2期)2019-07-08 03:19:59
大眾文藝(2019年10期)2019-06-05 05:55:32
百科知識(2016年22期)2016-12-24 21:07:25
當(dāng)代化工研究(2016年5期)2016-03-20 16:21:34
試題與研究·中考化學(xué)(2014年3期)2015-05-11 03:51:51
無機(jī)化學(xué)學(xué)報(2014年8期)2014-02-28 17:32:46
