
Structural characterization and comparative analysis of the chloroplast genome of Ginkgo biloba and other gymnosperms
2021-03-29 09:22:12XiaomingYangTingtingZhouXiyaSuGuibinWangXuhuiZhangQirongGuoFuliangCao
Journal of Forestry Research 2021年2期
Xiaoming Yang · Tingting Zhou · Xiya Su · Guibin Wang · Xuhui Zhang · Qirong Guo · Fuliang Cao
Abstract Ginkgo biloba is a famous living “fossil” and has played an important role in the evolution of the Plant Kingdom. Here, the complete chloroplast genome of G. biloba was sequenced and analysed. The chloroplast genome was 156,990 bp long and predicted to encode 134 genes including 85 protein-coding genes, 41 tRNA genes and 8 rRNA genes. The chloroplast genome has a typical quadripartite structure with a pair of inverted repeat regions (IRa and IRb, 17,732 bp), a large (LSC, 99,259 bp) and small single (SSC, 22,267 bp) copy region. After an extensive comparison to previously published gymnosperm plastomes, the gene content and organisation of G. biloba showed high divergence, although part was relatively conserved. The two typical IR regions in the G. biloba chloroplast genome were relatively shorter because it the ycf2 gene. In addition, it was obvious that the IR regions and gene loss were responsible for changes in chloroplast genome size and structure stability, which influenced plastome evolution in different gymnosperms. Phylogenetic analysis revealed that G. biloba is sister to cycads rather than to gnetophytes, cupressophytes, and Pinaceae. Overall, the study showed that the genomic characteristics of G. biloba would be of great help in the further research on the taxonomy, species identification and evolutionary history of gymnosperms, especially for their position in plant systematics and evolution.
Keywords Ginkgo biloba · Chloroplast genome · Comparative analysis · Phylogeny
Introduction
Ginkgo biloba L. (Ginkgoaceae) is called a “l(fā)iving fossil” and is a famous gymnosperm with edible seeds, ornamental value and medicinal properties (Lin et al. 2012). According to fossil evidence, the history of G. biloba dates back approximately to the early Permian (~ 280 Ma), and it is the sole surviving member of the genus Ginkgo, one of at least 16 genera of Ginkgoaceae (Gong et al. 2008). Only a few populations of G. biloba are endemic to an isolated location in China (Shen et al. 2005).
The chloroplast of plants and algae are double-membrane-bound organelles with their own genome that encode genes essential for photosynthesis and other metabolic pathways (Jansen and Ruhlman 2012). Generally, the chloroplast is originated from cyanobacteria through two independent secondary endosymbiosis and has its own genetic replication mechanism (Howe et al. 2003). The chloroplast genome is inherited maternally in the majority of angiosperms and gymnosperms, but in some gymnosperms, it is paternally inherited (Jansen and Ruhlman 2012). With the development of high-throughput sequencing technologies, the sequences of more and more chloroplast genomes have been released since the release of the first full chloroplast genomes sequences for liverwort and tobacco (Ohyama et al. 1986; Zheng et al. 2017). Increasingly, these chloroplast genomes have been widely used in molecular marker development, plant phylogeny evolution, DNA barcoding, population genetics and comparative genomics (Jansen and Ruhlman 2012; Yagi and Shiina 2014; Daniell et al. 2016; Sun et al. 2018; Hou et al. 2019).
Compared to plant nuclear and mitochondrial genomes, chloroplast genomes are typically small and less prone to recombination and provide distinct information about genome size variation and evolutionary status (Ravi et al. 2008; Jansen and Ruhlman 2012). The chloroplast genome consists of a relatively conserved quadripartite structure, usually harboring a pair of large inverted repeats (IRs; IRA and IRB), a prominent of feature of most land plant chloroplast genomes, dividing the genome into large (LSC) and small (SSC) single copy regions. The pairs of IRs are more likely to be influenced by the sequence length in both angiosperms and gymnosperms; in gymnosperms, the length of IRs fluctuates greatly among taxa (Guisinger et al. 2011; Jansen and Ruhlman 2012). Species of conifers and the legumes (Fabaceae) do not contain the large IR in the chloroplast genome, which lead to extensive genome sequence rearrangements (Wu et al. 2011). Genome rearrangements and more frequent gene loss and gain events are unique characteristics in most of gymnosperm chloroplast genomes. Species of Pinaceae and cupressophytes within conifers lost an IR copy in the chloroplast genomes (Wu and Chaw 2014), whereas both IRs have been retained in the chloroplast of Cycadales (Wu et al. 2007), Gnetales (Wu et al. 2009) and Ginkgoales (Lin et al. 2012), but their sizes variably changed.
In this study, we obtained a high quality, the complete chloroplast genome sequence for G. biloba by high-throughput sequencing. Using comparative genomics and four previously published genome sequences for gymnosperm chloroplasts, we characterized the chloroplast genome structure of G. biloba and brought deeper insights into the evolutionary aspects of its chloroplast genome. The results provide a theoretical basis for future research.
Materials and methods
DNA sequencing, chloroplast genome assembly, and validation
Fresh leaves were collected from a female tree of G. biloba at Nanjing Forestry University (Jiangsu Province, P. R. China), and total DNA was isolated using the DNeasy Plant Mini Kit (Germany, Qiagen). Specifically, pure DNA was used to construct shotgun library (250 bp) according to the requirements by the Illuminate Hiseq 2500 platform (Allwegene, China). Approximately 300 Gb of raw data were obtained from the sample.
The complete G. biloba chloroplast genome was assembled using bioinformatic pipelines (Wang et al. 2018) with some modifications. First, low-quality reads (Phred scores < 30) were removed from the total raw reads using Trimmomatic version 0.36 (Bolger et al. 2014). High-quality clean reads were then aligned against the published sequence of G. biloba chloroplast genome (Lin et al. 2012) through BLASTN search (E-value 1e?5 and -word_size 9) to extract cp-like reads. Then, these cp-like reads were assembled into contigs using the ABySS version 2.0 (Jackman et al. 2017) with k = 45. The CD-Hit software (? c 0.8 ? aS 0.8 ? d 0) (Fu et al. 2012) was used to remove redundant contigs and merge unique contigs. Sequence extension was executed using SSPACE (? x 1 ? r 0.9 ? a 0.7) (Boetzer et al. 2011), and gaps were filled using CapCloser version 1.12 (? m 100) (Luo et al. 2012). The adjacent regions of sequences between IRs and LSC/SSC in the chloroplast genome were needed to confirm by polymerase chain reaction amplification based on Sanger sequencing with primers described in Table S1. The detailed amplification reaction and sequencing of chloroplast DNA were done according to our previously described protocol (Yang et al. 2015). Finally, Short reads from Illumina sequencing were then mapped to preliminary assembled reference genomes with BWA version 0.7.16a (r1181) (aln ? t 30 ? f) (Li and Durbin 2009), forming consensus sequences to further determine whether there were base differences in the reference.
Chloroplast genome annotation and sequence analyses
Preliminary genome annotation was done using CpGAVAS2 (Shi et al. 2019) and coupled with adjustment in GeSeq (Tillich et al. 2017). All tRNA genes were identified by tRNAscan-SE version 1.21 (Schattner et al. 2005). Circular gene maps were drawn using the online program OGDRAW version 1.2 (Wyman et al. 2004) followed by manual checking.
Comparative genome analyses
Cycads, ginkgo, gnetophytes, Pinaceae (conifers I) and cupressophytes (conifers II) totally constituted the five major groups in gymnosperm (Jansen and Ruhlman 2012). We selected ginkgo and other four representative species [ Pinus taeda (KY964286), Taxus chinensis var. Mairei (KJ123824), Cycas debaoensis (KU743927) and Gnetum montanum (NC_021438)] from each different group in gymnosperms to elucidate the chloroplast genome size and structure variations. Pairwise sequence alignments of different chloroplast genomes were done using the program MUMmer (Kurtz et al. 2004). We used the program mVISA (Mayor et al. 2000) to compare the chloroplast genome of G. biloba with the other four gymnosperms in the shuffl e-LAGAN mode using the annotation of G. biloba as a reference. Circular genome visualisation and exploration among these five genomes were done using CGView (Grant and Paul 2008). Variations in AT content, codon usage, synonymous codon usage and RSCU were determined using MEGA version 7.0 (Kumar et al. 2016).
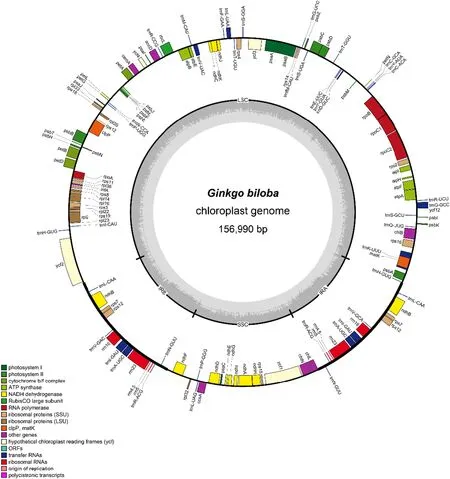
Fig.1 Chloroplast genome maps of G. biloba. Genes inside the circle are transcribed and color-coded by functional group. Light gray shows AT content; dark gray in the inner circle shows GC content
Elucidation of repeat sequences and cpSSRs
Forward, reverse and palindromic repeats were identified within the G. biloba chloroplast genome using the program REPuter (Kurtz et al. 2001). The parameters were set as follows: the minimum repeat size of sequence was 20 bp and the sequence identity was more than 90% (hamming distance equal to 3). Furthermore, the MISA software (Thiel et al. 2003) was used to exploit potential cpSSRs with the G. biloba chloroplast genome sequence. To compare the characteristics of repeat sequences and SSRs of G. biloba to those of P. taeda, T. chinesis, C. debaoensis and G. montanum, these four gymnosperm chloroplast genomes were also analyzed using the same methods above.
Phylogenetic analysis
All 38 shared protein-coding genes (Table S2) of 22 species from five families of gymnosperms and 2 species of angiosperms (Table S3) were used in alignment analyses with ClustalW (Thompson et al. 1994). The alignment results were checked manually. The general model for nucleotide sequence (GTR + G + I) was conducted using jModeltest 2 (Darriba et al. 2012) with optimized parameters. Optimal phylogenetic trees were constructed using MP and ML as implemented in PAUP* version 4.0b10 (Swofford 2002) and RAxML version 8.1.5 (Stamatakis 2014) with 1000 bootstrap replicates. Characters were equally weighted, gap regions were excluded and multistate characters were treated as uncertainties for maximum parsimony (MP) and maximum likelihood (ML) analyses. In addition, both Arabidopsis thaliana and Poplus euphratica were set as outgroups.
Results and discussion
Features of G. biloba chloroplast
We assembled the chloroplast genome of G. biloba and submitted to the NCBI database (GenBank accession number MN443423). Notably, the chloroplast genome of G. biloba had a quadripartite structure that contained a pair of IR regions (17,732 bp), LSC region (99,259 bp) and SSC region (22,267 bp) (Fig.1; Table 1). The chloroplast genome of G. biloba was 156,990 bp in size, which was smaller than that of C. debaoensis (162,094 bp) (Jiang et al. 2016) but larger than that of T. chinensis (129,513 bp) (Zhang et al. 2014), P. taeda (121,531 bp) (Asafet al. 2018) and G. montanum (115,019 bp) (Mao et al. 2017). Two typical IR regions were found in G. biloba, C. debaoensis , and G. montanum, but
T. chinensis and P. taeda just retained one part of the IR regions. The IRs of ginkgo did not include ycf 2 (6540 bp), which caused the chloroplast genome size to be smaller than that of cycads. Therefore, there is reason to believe that the chloroplast genome size varied greatly in gymnosperms due to unstable genome structure (Wu et al. 2009) or loss of IRs or genes (Lin et al. 2012).
The number of genes in plant chloroplast genomes ranges from 63 to 209 but is concentrated between 110 and 130, with a conserved composition and arrangement (Jansen and Ruhlman 2012). A total of 134 genes were identified in the G. biloba chloroplast genome, including 85 protein-coding genes, 8 ribosomal RNA (rRNA) genes, and 41 transfer RNA (tRNA) genes (Fig.1; Table 2). Among these genes, 12 tRNA genes, 8 rRNA genes, and 4 protein genes are duplicated in the IR regions (Fig.1). There were 66 protein-coding and 27 tRNA genes in the LSC region, whereas the SSC region included 14 protein-coding genes and 2 tRNA genes. In total, there were 21 intron-containing genes (Table 3), 23 (8 tRNA genes and 13 protein-coding genes) of which contained one intron, and 2 ( clpP and ycf3) of which contained 2 introns. The matk gene located in the intron of the trnK-UUU gene. Gene rps 12 was a trans-spliced gene and the 5′and 3′ ends of rps12 were distributed in the LSC and IR regions, respectively. The ycf3 gene in G. biloba harbored two introns, which would promote further researches about the mechanism of photosynthesis evolution because studies have shown that ycf3 is necessary for the stable accumulation of the photosystem I complex (Boudreau et al. 1997) and that introns in a gene can affect the gene expression (Shaul 2017).

Table 1 Summary of chloroplast genome features of five gymnosperms
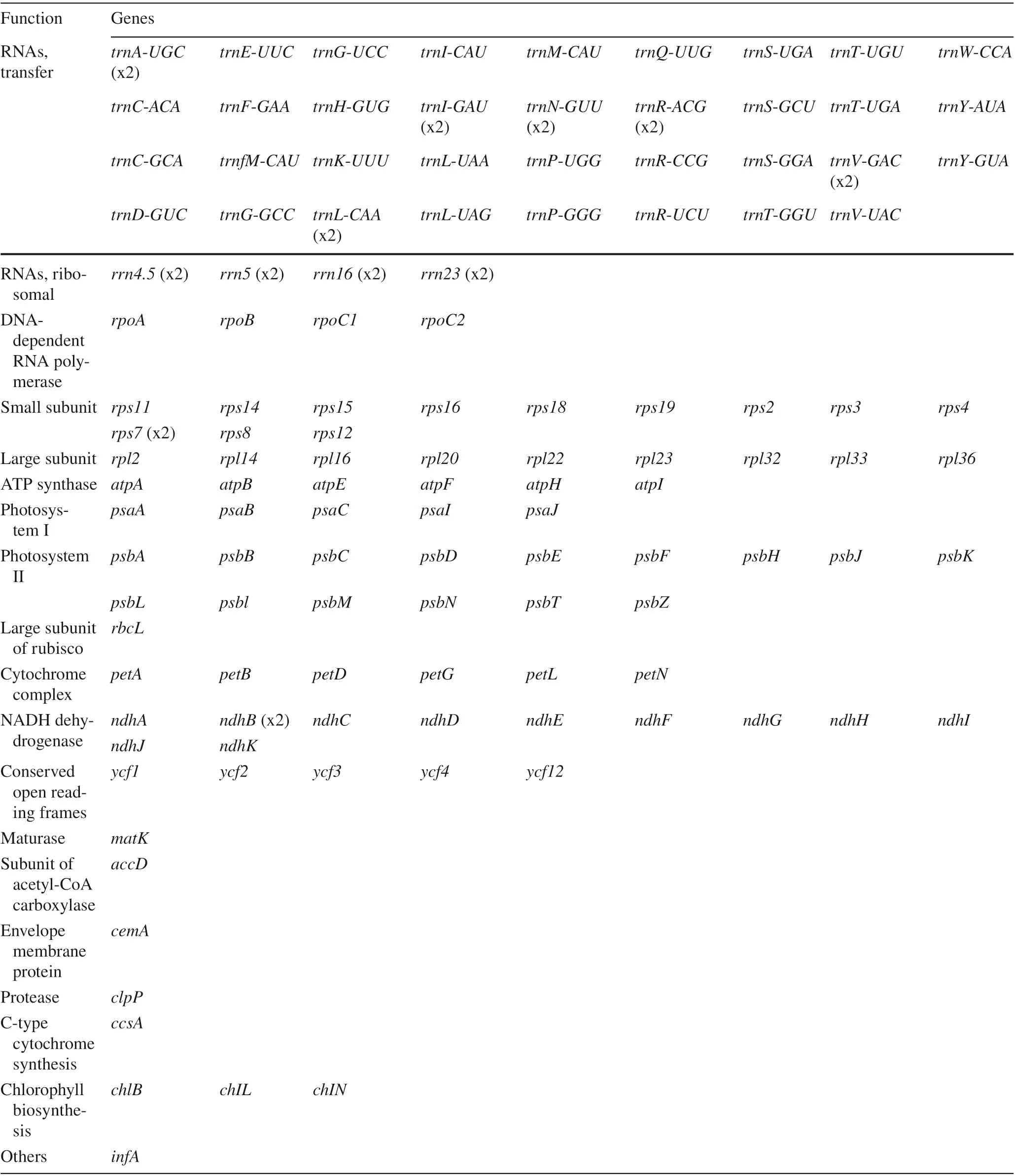
Table 2 List of genes annotated in the chloroplast genome of G. biloba
Generally, the GC base pairs were more thermodynamically stable than AT base pairs, which led to the GC content having a more significant influence on the chloroplast genome stability. The GC content of the LSC, SSC, and two IR regions in the chloroplast genome of G. biloba, was 38.5%, 36.2%, 44.6%, and 44.6%, respectively (Table 4).The GC content of the whole chloroplast in G. biloba was similar to that ofother three gymnosperms except for T. chinensis. Interestingly, the GC content of the IR regions was higher than that of the other regions in G. biloba, C. debaoensis and G. montanum, a common phenomenon in chloroplast genomes and that attributable to the distribution of the rRNA and tRNA genes in relatively shorter genome regions (Guo et al. 2018). Apart from the abovementioned gymnosperms, these patterns are also similar to those in other gymnosperms (Wu and Chaw 2014). Furthermore, the AT content of G. biloba chloroplast protein-coding regions at the first, second and third codon positions was 52.97%, 60.52% and 69.62%, respectively. The third codon position tends to have higher AT content, which may be regarded as a common feature in most of plant chloroplast genomes (Nie et al. 2012; Chen et al. 2015; Li et al. 2018).

Table 3 Location and length of intron-containing genes within the G. biloba chloroplast genome
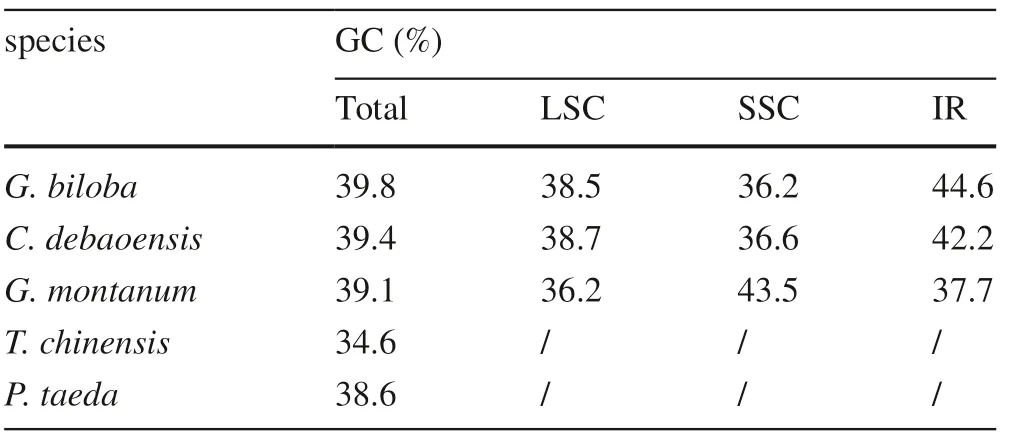
Table 4 GC content of large single copy (LSC) region, small single copy (SSC) region and inverted repeats (IRs) in five gymnosperms
Twenty amino acids that could be transported for protein biosynthesis by tRNA molecules in G. biloba plastome. A total of 27,559 codons were identified in all combined coding sequence (CDS) in the G. biloba plastome. The frequency of the relative synonymous codon usage (RSCU) was estimated for the G. biloba chloroplast genome (Fig.2). Among all codons, 2962 (10.74%) codons encoded for phenylalanine (the most abundant), and 341 (1.24%) codons encoded for cysteine (least abundant). The RSCU value was consistent with the number of codons encoding a particular amino acid. Except for methionine and tryptophan, most amino acid codons exhibited preferential use, and codons ending with A or U were common.
Long-repeat and simple sequence repeat (SSR) analysis
Numerous tandem repeats are associated with gene duplication, rearrangement and gene expansion in many species (Marechal and Brisson 2010). For the repeat structure analysis, the G. biloba chloroplast genome contained 22 forward repeats, 26 inverted repeats and 1 palindromic repeat (Fig.3), and 49, 50, 50, and 50 repeat pairs were found in the other four represented gymnosperm chloroplast genome, T. chinesis, P. taeda, G. montanum, and C. debaoensis, respectively (Fig.3). The length of most forward and reverse repeats ranged between 20 and 34 bp, whereas the number of palindromic repeats was limited, and there was only one palindromic repeat in G. biloba (Fig.3). Although the total number of repeats was similar among the five species, the proportion of three repeat types in each species showed a significant difference (sequence number and length), indicating that each species had a different evolutionary history. Variation in these repeat motifs is an informative source for developing markers and appropriate for population genetics and evolutionary studies (Nie et al. 2012; Chen et al. 2015).
The number of chloroplast simple sequence repeats (cpSSRs) in G. biloba (37) chloroplast genome was similar to that in C. debaoensis (40) and G. montanum (40) but higher than in T. chinensis (24) and P. taeda (24) chloroplast genomes (Fig.4 ). Among these SSRs, six of them were located in genes, and the remaining genes were distributed in the intergenic regions in the G. biloba chloroplast genome. Although the length of the G. montanum chloroplast genome was shortest among these five species, the number of SSRs was obviously greater than in T. chinensis and P. taeda, the two species that had a relatively small chloroplast genomes size. Therefore, longer chloroplast genome sequences do not necessarily have more SSRs. The mononucleotide and dinucleotide repeats were the most frequent types of SSR in G. biloba, especially the mononucleotide repeat content, which accounted for the largest proportion (67.57%). We also found a similar phenomenon in the other four gymnosperms, in which the mononucleotide was the most common type of SSR, and the proportion of the mononucleotide repeat content was 58.33% in T. chinesis, 79.17% in P. taeda, 90% in G. montanum and 85% in C. debaoensis. In addition, 96% of the mononucleotide SSRs constituted A/T sequences, whereas only one belonged to the G/C motif. Interestingly, 87.50% of the dinucleotide SSRs were also A/T motifs. Most of the cpSSRs were composed of short polyadenine (poly A) or polythymine (poly T) repeats instead of tandem guanine (G) or cytosine (C). Compared to other neutral regions, SSRs usually have a higher rate of mutation due to slipped-strand mispairing (Wheeler et al. 2014). For high variability at the intraspecific level, cpSSRs are very useful in population genetics and evolutionary studies because these short repeats have haploid and uniparentally inherited characteristics (Wheeler et al. 2014). Thus, cpSSR markers developed in the G. biloba chloroplast genome could be used to evaluate genetic diversity and potentially to distinguish different germplasms.
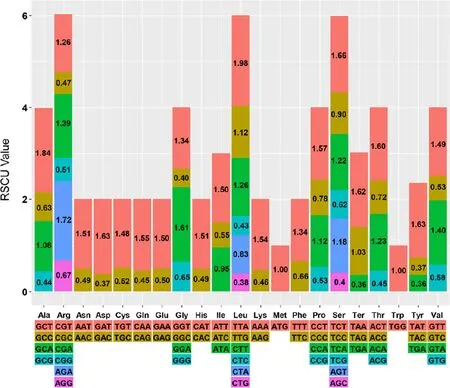
Fig.2 Codon content for 20 amino acids and stop codons in all protein-coding genes in the G. biloba chloroplast genome
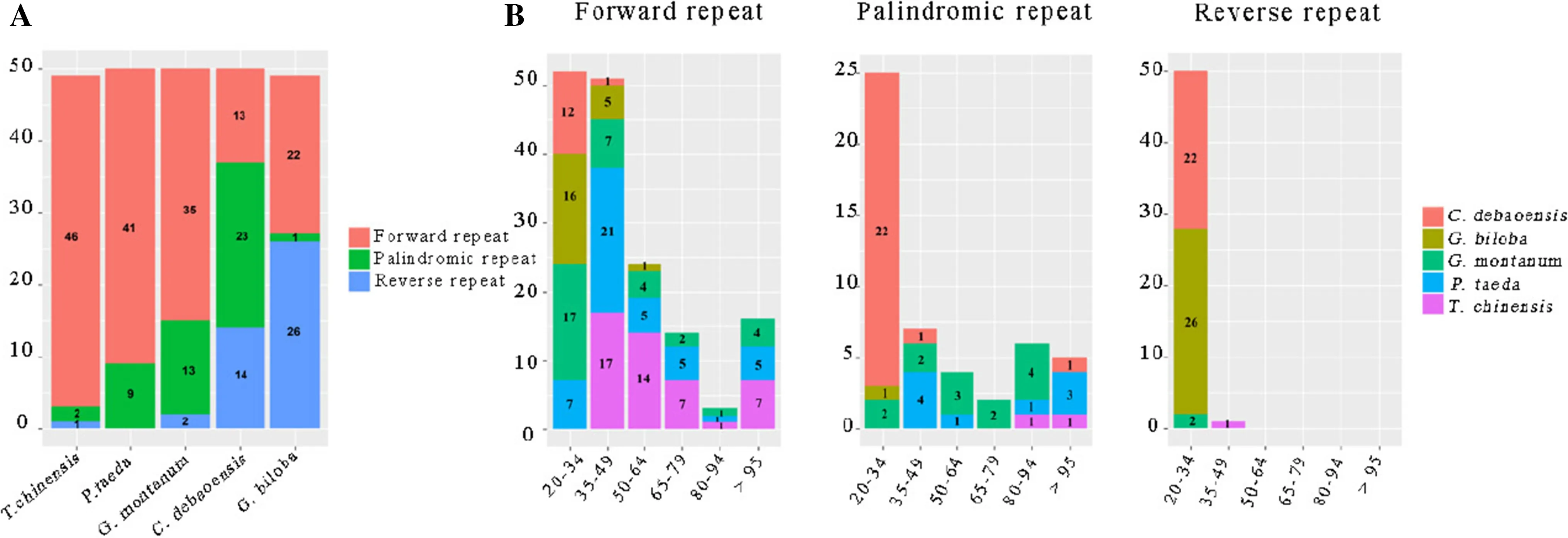
Fig.3 Repeated sequences in five gymnosperm chloroplast genomes. a Totals of three kinds of repeat types; b frequency of forward, palindromic and tandem repeats by length
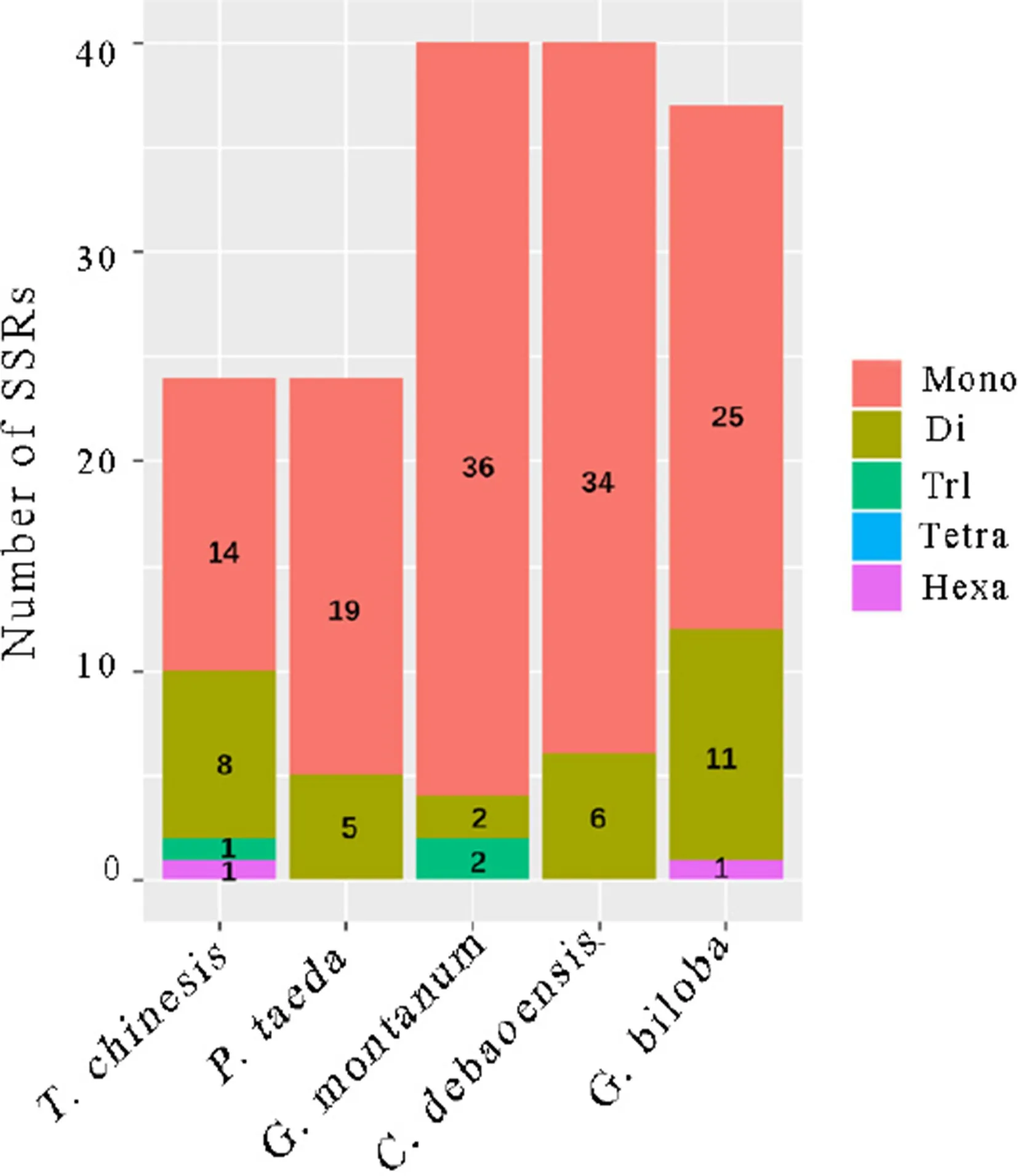
Fig.4 Analysis of simple sequence repeats (SSRs) from five different gymnosperm chloroplast genomes
Comparative chloroplast genomic analysis among the five gymnosperms
The comparative analyses of chloroplast genomes can elucidate the complex evolutionary history of species and is an extremely important step in phylogenomics (Zheng et al. 2017). According to the previous researches, three important factors including gene loss, IR and intergenic region variations, significantly influences on the chloroplast genome size and structure in plants (Daniell et al. 2016; Chaw et al. 2018). The chloroplast genome size, genome structure and gene numbers of gymnosperm can vary much more than those of angiosperms because species in gymnosperm have complex evolutionary histories and genetic backgrounds (Moore et al. 2007; Chaw et al. 2018). When we compared G. biloba to the other four representative gymnosperms species, the results clearly demonstrated that high divergence among most of the aligned chloroplast genome sequences, although part was relatively conserved (Figs. 5 and 6; Fig.S1). The most remarkable characteristic of plastome size variation within these five species occurred in G. montanum that had the smallest plastome. The elimination of dispensable sequences in G. montanum cpDNA is mainly due to segment deletions, which is more effi cient than single nucleotide deletions (Gregory 2004). A compact cpDNA may be due to undergo selection for a reduced genome (Wu et al. 2009). Therefore, there would be a strong deletion for the compaction of the gnetophyte chloroplast genome. Meanwhile, we also observed that G. montanum had contracted plastomes from the downsizing of introns and intergenic spacer regions. Similarly, T. chinesis cpDNAs had evolved towards reduced size, also largely due to shrunken intergenic spacers.
Most land plants have IRs that have played an important role in plastome evolution. The IRs of the five gymnosperm different groups have gone through distinctive evolutionary histories; thus, the IR regions are one of the most obvious differences in chloroplast genome. Losing or retaining both or either of the IR regions influence the stability of genomic structure and more rearrangements are more likely to occur in the chloroplast genome that lost the IRs than that contained the IRs (Hirao et al. 2008). Detailed comparisons of the IR-SSC and IR-LRC boundaries among the chloroplast genomes of five gymnosperms were presented in Fig.7. The IR regions were the least conservative among G. biloba, C. debaoensis and G. montanum that contained two typical IR regions, not to mention T. chinesis and P. taeda that just contained one incomplete IR region (Fig.7). Except for tribes of the legume family (Fabaceae), it is a rare occurrence that species lost their IRs in angiosperms (Jansen and Ruhlman 2012). However, the absence of an IR copy is a unique characteristic shared by almost all conifers, which is considered to be a derived factor that influenced gene lose-and-gain events and genomic rearrangements in chloroplast genome of cupressophytes and Pinceae (Wu et al. 2007). The putative ancestral IRs of gymnosperms comprise 15 genes (Zhu et al. 2016), which were all retained in the IRs of C. debaoensis, but that of G. biloba just contained 13 genes, excluding ycf 2 in the IRs. The LSC-IR junction region retained the complete ycf2 and the adjoining psbA or rpl 23- rps3 gene cluster, which would be regarded as ancestral IRs because the genes in the LSC-IR junction region are usually in order (Wu et al. 2011). Thus, we reasonably speculate that a different copy of IRs was lost in P. taeda and T. chinesis, with the former losing IRB and the latter losing IRA. Beyond that, however, the pair of IRs in the plastomes of P. taeda reduced extremely and just included trnI- CAU and 3′ psbA. Therefore, it was more diffi cult to accurately identify IR residues for P. taeda. Although G. biloba, G. montanum and C. debaoensis retained two typical IR regions, G. biloba showed a specific genome structure characteristic in IR regions. The shortened IR of ginkgo cpDNA among these three species was a consequence of IR contraction, which was mainly due to the loss ofone ycf2 copy from the IR region. Therefore, there is no doubt that sequence rearrangements changed LSC and SSC regions in gymnosperm chloroplast genome structure, which delivered genetic diversity information to carry out studies on molecular classification and phylogeny.

Fig.5 Structure of five gymnosperm chloroplast genomes constructed using mVISTA, with the G. biloba genome as the reference. Vertical and horizontal axes represent the degree of consistency among the sequences from (50–100%) and the sequence lengths, respectively. Exons, untranslated regions (UTRs), noncoding sequences (CNS) and mRNA are color-coded. Annotated genes are displayed along the top
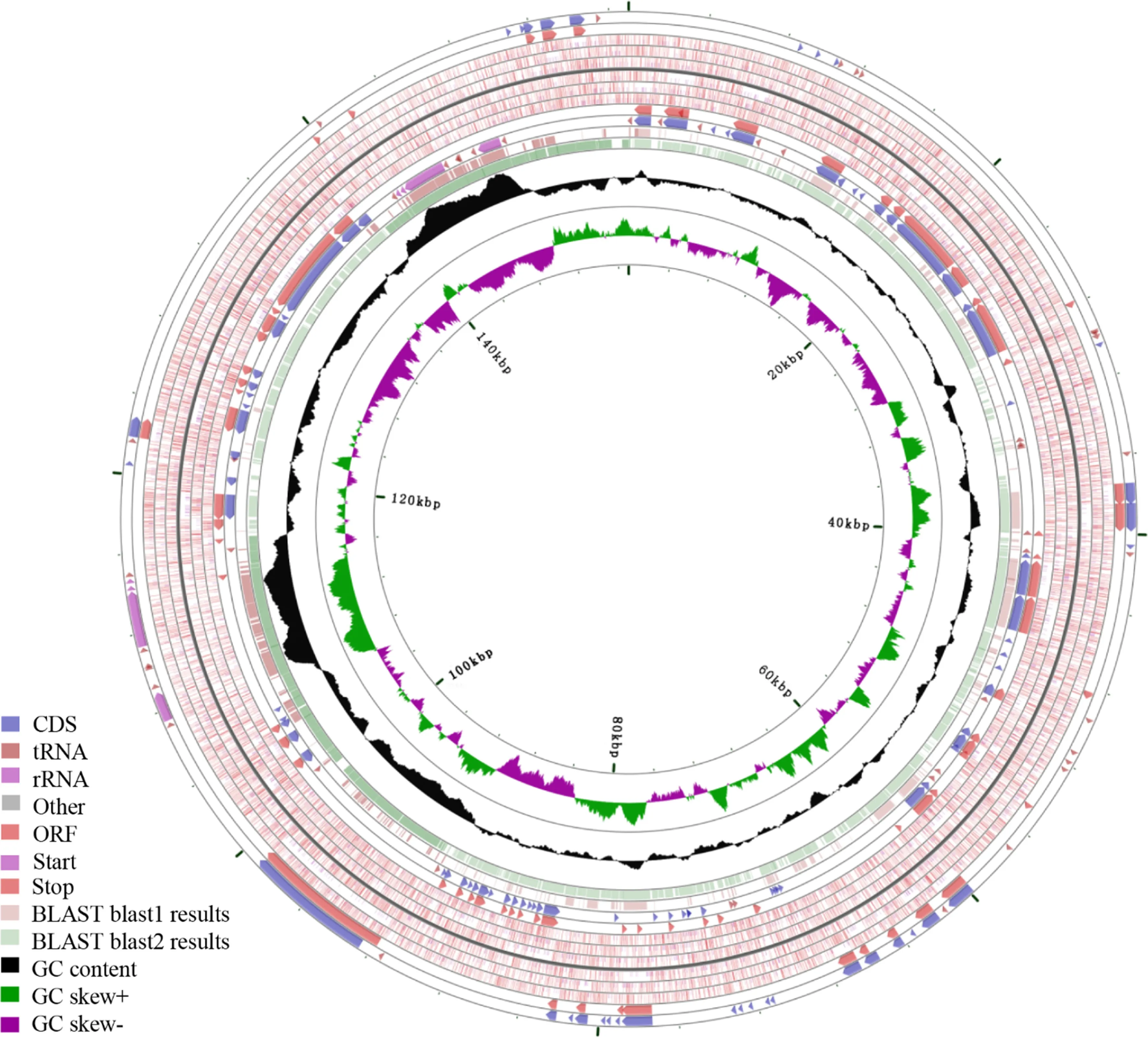
Fig.6 Comparison of chloroplast genomes from three gymnosperms using GCview. The outer two rings pointing in different directions show the coding sequence, rRNA genes, and tRNA genes. The two inner circles show the blast results for G. biloba vs G. montanum, and C. debaoensis, respectively. GC skew + (green) means G > C, whereas GC skew- (purple) indicates G < C
Phylogenetic analysis
Holding great potential for the studies of phylogenetics, evolution and molecular systematics, chloroplast genomes have been widely used to solve phylogenetic questions in many land plants (Zheng et al. 2017). Phylogenetic relationships among the gnetophytes, cupressophytes, cycads, ginkgo, and Pinaceae, which are closely related to ginkgo, have been hotly debated for many years. For understanding the phylogenetic relationship of G. biloba within the gymnosperms, the 38 protein-coding genes shared in the chloroplast genome of 22 gymnosperms and 2 angiosperms ( A. thaliana and P. euphratica as outgroups) were selected. After successfully concatenating the 38 protein-coding genes, we constructed an evolutionary tree based on the ML and MP analysis. Bootstrap values of almost all phylogenetic relationships inferred from the chloroplast protein gene data were generally high based on the MP and ML methods (the support values ranged from 0.98 to 1).
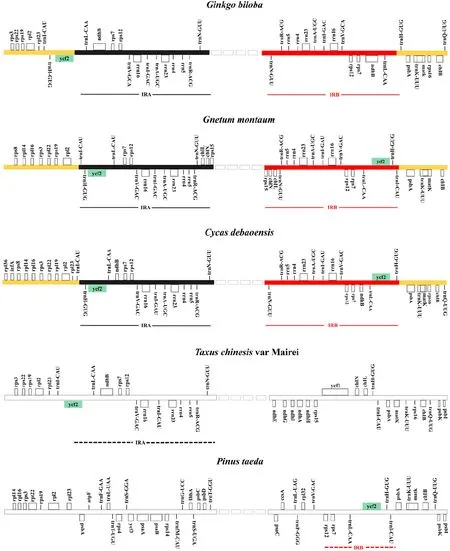
Fig.7 Comparison of IR boundaries among five gymnosperms ( G. biloba, C. debaoensis, G. montanum, T. chinesis and P. taeda) revealed different IR copies retained. G. biloba, C. debaoensis and G. montanum had integrated pairs of IRs. However, T. chinesis had only IRA region and P. taeda retained part of IRB region
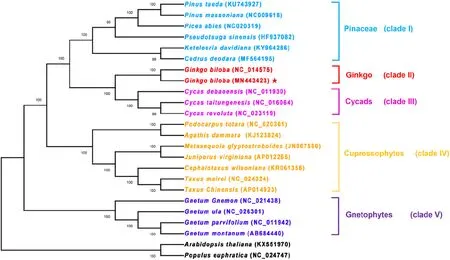
Fig.8 Phylogenetic relationships among 24 species inferred using maximum likelihood analyses of the shared 38 shared protein-coding genes in the chloroplast genome. Numbers at nodes are values for bootstrap support. GenBank accessions follow each species in the brackets; chloroplast genome of G. biloba sequenced here is marker by a red star. Sequences from A. thaliana and P. euphratica served as outgroups
It was noteworthy that the ML (Fig.8) and MP (Fig.S2) trees were similar phylogenetic topologies, with six clades: Pinaceae (conifer I), cupressophytes (conifer II), cycads, gnetophytes, ginkgo and the out group. Interestingly, both MP and ML phylogenetic analyses showed strong support that G. biloba was distant from cupressophytes, gnetophytes and Pinaceae but rather much closer to the cycads with a high bootstrap value (100% in the MP and 100% in ML tree). Both of nuclear phylotranscriptomic and nuclear phylogenomic analyses also support that ginkgo is a sister to the cycads (Wickett et al. 2014; Li et al. 2017). The rest of the gymnosperms formed three relatively independent evolution branches. Among these species, the first clade (clade I) contained six species that belong to Pinaceae (conifer I), and clade IV comprised all investigated cupressophytes. Within clade V, all species were gnetophytes. Furthermore, the phylogenetic topology of gymnosperms with chloroplast genes in our research is congruent with previous phylogenetic analyses (Wu et al. 2011, 2013; Chen et al. 2015). Thus, the MP and ML phylogenetic results strongly supported that gnetophytes were at the base of gymnosperms tree and that cycads were the sister species to ginkgo rather than to gnetophytes, cupressophytes and Pinaceae.
Conclusions
Here we present the complete chloroplast genome of G. biloba, an endemic and ecological tree in China. From our characterisation of the chloroplast genome structural organisation and comparative analyses of the chloroplast genomes of G. biloba and other gymnosperms, we found that the chloroplast genome of G. biloba had a typical quadruple structure and the gene order, GC content and codon usage features differed from the other gymnosperm chloroplast genomes. Variation in IRs and gene loss were the main factors influencing chloroplast genome size and structural stability in the five gymnosperms. The chloroplast genome structure of G. biloba contained a shortened IR resulting from the IR contraction, and the contraction mainly resulted from the loss of the ycf 2 copy from the IRs. Phylogenetic relationships strongly supported that ginkgo had a closer relationship with cycads than with gnetophytes, Pinaceae and cupressophytes. Our results showed the clear chloroplast genome structural characterisation of G. biloba, which lays a foundation for future breeding and further biological discoveries.
AcknowledgementsThe authors sincerely thank laboratory members for assistance with the study.
References
Asaf S, Khan AL, Khan MA, Shahzad R, Lubna Kang SM, Al-Harrasi A, Al-Rawahi A, Lee IJ (2018) Complete chloroplast genome sequence and comparative analysis of loblolly pine ( Pinus taeda L.) with related species. PLoS ONE 13(3):e0192966
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W (2011) Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27(4):578–579
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120
Boudreau E, Takahashi Y, Lemieux C, Turmel M, Rochaix JD (1997) The chloroplast ycf3 and ycf4 open reading frames of Chlamydomonas reinhardtii are required for the accumulation of the photosystem I complex. EMBO J 16(20):6095–6104
Chaw SM, Wu CS, Sudianto E (2018) Evolution of gymnosperm plastid genomes. In: Advances in botanical research. Elsevier, New York, pp 195–222
Chen JH, Hao ZD, Xu HB, Yang LM, Liu GX, Sheng Y, Zheng C, Zheng WW, Cheng TL, Shi JS (2015) The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front Plant Sci 6:447
Daniell H, Lin CS, Yu M, Chang WJ (2016) Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 17(1):134
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models new heuristics and parallel computing. Nat Methods 9(8):772
Fu LM, Niu BF, Zhu ZW, Wu ST, Li WZ (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28(23):3150–3152
Gong W, Zeng Z, Chen YY, Chen C, Qiu YX, Fu CX (2008) Glacial refugia of Ginkgo biloba and human impact on its genetic diversity: evidence from chloroplast DNA. J Integr Plant Biol 50(3):368–374
Grant JR, Paul S (2008) The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res 36:181–184
Gregory TR (2004) Insertion-deletion biases and the evolution of genome size. Gene 324:15–34
Guisinger MM, Kuehl JV, Boore JL, Jansen RK (2011) Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol 28(1):583–600
Guo S, Guo L, Zhao W, Xu J, Li YY, Zhang XY, Shen XF, Wu ML, Hou XG (2018) Complete chloroplast genome sequence and phylogenetic analysis of Paeonia ostii. Molecules 23:246
Hirao T, Watanabe A, Kurita M, Kondo T, Takata K (2008) Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: diversified genomic structure of coniferous species. BMC Plant Biol 8:70
Hou Z, Wang ZS, Zhang JG (2019) The complete chloroplast genomic landscape and phylogenetic analysis of Populus alba L. J For Res. https://doi.org/10.1007/s1167 6-019-00953-6
Howe CJ, Barbrook AC, Koumandou VL, Nisbet RER, Symington HA, Wightman TF (2003) Evolution of the chloroplast genome. Philos Trans R Soc Lond B Biol Sci 358(1429):99–107
Jackman SD, Vandervalk BP, Mohamadi H, Chu J, Yeo S, Hammond SA, Jahesh G, Khan H, Coombe L, Warren RL (2017) ABySS 2.0: Resource-effi cient assembly of large genomes using a Bloom filter. Genome Res 27(5):768–777
Jansen RK, Ruhlman TA (2012) Plastid genomes of seed plants. In: Bock R, Knoop V (eds) Genomics of chloroplasts and mitochondria. Springer, Dordrecht, p 377
Jiang GF, Hinsinger DD, Strijk JS (2016) Comparison of intraspecific, interspecific and intergeneric chloroplast diversity in Cycads. Sci Rep 6:31473
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 70 for bigger datasets. Mol Biol Evol 33(7):1870–1874
Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R (2001) REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res 29(22):4633–4642
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL (2004) Versatile and open software for comparing large genomes. Genome Biol 5(2):12
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760
Li Z, De La Torre AR, Sterck L, Canovas FM, Avila C, Merino I, Cabezas J, Cervera M, Ingvarsson PK, Van de Peer Y (2017) Single-copy genes as molecular markers for phylogenomic studies in seed plants. Genome Biol 9(5):1130–1147
Li X, Li YF, Zang MY, Li MZ, Fang YM (2018) Complete chloroplast genome sequence and phylogenetic analysis of Quercus acutissima. Int J Mol Sci 19:2443
Lin CP, Wu CS, Huang YY, Chaw SM (2012) The complete chloroplast genome of Ginkgo biloba reveals the mechanism of inverted repeat contraction. Genome Biol Evol 4(3):374–381
Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, Yuan JY, He GZ, Chen YX, Pan Q, Liu YJ, Tang JB, Wu GX, Zhang H, Shi YJ, Liu Y, Yu C, Wang B, Lu Y, Han CL, Cheung DW, Yiu SM, Peng SL, Zhu XQ, Liu GM, Liao XK, Li YR, Yang HM, Wang J, Lam TW, Wang J (2012) SOAPdenovo2: an empirically improved memory-effi cient short-read de novo assembler. Gigascience 1(1):18
Mao JP, Zhou F, Liu TY, Wu ZY, Zhong T, Liu CX, Wei Q, Chen JH, Huang SW (2017) The complete chloroplast genome of Gnetum montanum and sequence analysis. Mitochondrial DNA A 28(3):409–410
Marechal A, Brisson N (2010) Recombination and the maintenance of plant organelle genome stability. Int J Mol Sci 186:299–317
Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I (2000) VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 16(11):1046–1047
Moore MJ, Bell CD, Soltis PS, Soltis DE (2007) Using plastid genomescale data to resolve enigmatic relationships among basal angiosperms. Proc Natl Acad Sci USA 104(49):19363–19368
Nie XJ, Lv SZ, Zhang YX, Du XH, Wang L, Biradar SS, Tan XF, Wan FH, Song WN (2012) Complete chloroplast genome sequence of a major invasive species, crofton weed ( Ageratina adenophora). PLoS ONE 7(5):e36869
Ohyama K, Fukuzawa H, Kohchi T, Shirai H, Sano T, Sano S, Umesono K, Shiki Y, Takeuchi M, Chang Z (1986) Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322:572–574
Ravi V, Khurana JP, Tyagi AK, Khurana P (2008) An update on chloroplast genomes. Plant Syst Evol 271(1–2):101–122
Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res 33(2):686–689
Shaul O (2017) How introns enhance gene expression. Int J Biochem Cell Biol 91:145–155
Shen L, Chen XY, Zhang X, Li YY, Fu CX, Qiu YX (2005) Genetic variation of Ginkgo biloba L. (Ginkgoaceae) based on cpDNA PCR-RFLPs: inference of glacial refugia. Heredity 94:396–401
Shi LC, Chen HM, Jiang M, Wang LQ, Wu X, Huang LF, Liu C (2019) CPGAVAS2 an integrated plastome sequence annotator and analyzer. Nucleic Acids Res 47(1):65–73
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313
Sun CR, Li J, Dai XG, Chen YN (2018) Analysis and characterization of the Salix suchowensis chloroplast genome. J For Res 29(4):1003–1011
Swofford DL (2002) PAUP: phylogenetic analysis using parsimony version 40 b10. Sinauer Associates, Sunderland
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of genederived SSR-markers in barley ( Hordeum vulgare L.). Theor Appl Genet 106(3):411–422
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Bock R, Greiner S (2017) GeSeq-versatile and accurate annotation oforganelle genomes. Nucleic Acids Res 45(1):6–11
Wang XL, Cheng F, Rohlsen D, Bi CW, Wang CY, Xu YQ, Wei SY, Ye QL, Yin TM, Ye N (2018) Organellar genome assembly methods and comparative analysis of horticultural plants. Hortic Res 5(1):3
Wheeler GL, Dorman HE, Buchanan A, Challagundla L, Wallace LE (2014) A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl Plant Sci 2(12):1400059
Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA, Ruhfel BR, Wafula E, Der JP, Graham SW, Mathews S, Melkonian M, Soltis DE, Soltis PS, Miles NW, Rothfels CJ, Pokorny L, Shaw AJ, DeGironimo L, Stevenson DW, Surek B, Villarreal JC, Roure B, Philippe H, dePamphilis CW, Chen T, Deyholos MK, Baucom RS, Kutchan TM, Augustin MM, Wang J, Zhang Y, Tian Z, Yan Z, Wu X, Sun X, Wong GK-S, Leebens-Mack J (2014) Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc Natl Acad Sci USA 111(45):4859–4868
Wu CS, Chaw SM (2014) Highly rearranged and size-variable chloroplast genomes in conifers II clade (cupressophytes): evolution towards shorter intergenic spacers. Plant Biotechnol J 12(3):344–353
Wu CS, Wang YN, Liu SM, Chaw SM (2007) Chloroplast genome (cpDNA) of Cycas taitungensis and 56 cp protein-coding genes of Gnetum parvifolium: insights into cpDNA evolution and phylogeny ofextant seed plants. Mol Biol Evol 24(6):1366–1379
Wu CS, Lai YT, Lin CP, Wang YN, Chaw SM (2009) Evolution of reduced and compact chloroplast genomes (cpDNAs) in gnetophytes: selection toward a lower-cost strategy. Mol Phylogenet Evol 52(1):115–124
Wu CS, Wang YN, Hsu CY, Lin CP, Chaw SM (2011) Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and Cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biol Evol 3:1284–1295
Wu CS, Chaw SM, Huang YY (2013) Chloroplast phylogenomics indicates that Ginkgo biloba is sister to cycads. Genome Biol Evol 5(1):243–254
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation oforganellar genomes with DOGMA. Bioinformatics 20(17):3252–3255
Yagi Y, Shiina T (2014) Recent advances in the study of chloroplast gene expression and its evolution. Front Plant Sci 5:61
Yang XM, Li H, Liang M, Xu Q, Chai LJ, Deng XX (2015) Genetic diversity and phylogenetic relationships of citron ( Citrus medica L) and its relatives in southwest China. Tree Genet Genomes 11(6):129
Zhang YZ, Ma J, Yang BX, Li RY, Wei Z, Sun LL, Tian JK, Zhang L (2014) The complete chloroplast genome sequence of Taxus chinensis var. mairei (Taxaceae): loss of an inverted repeat region and comparative analysis with related species. Gene 540(2):201–209
Zheng XM, Wang JR, Li F, Sha L, Pang HB, Lan Q, Jing L, Yan S, Qiao W, Zhang LJSR (2017) Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci Rep 7:1555
Zhu AD, Guo WH, Gupta S, Fan WS, Mower JP (2016) Evolutionary dynamics of the plastid inverted repeat: the effects ofexpansion, contraction, and loss on substitution rates. New Phytol 209:1747–1756
 Journal of Forestry Research2021年2期
Journal of Forestry Research2021年2期
- Journal of Forestry Research的其它文章
- Stem taper functions for Betula platyphylla in the Daxing’an Mountains, northeast China
- Leaf phenotypic variation of endangered plant Tetracentron sinense Oliv. and influence of geographical and climatic factors
- Ecological variations of woody species along an altitudinal gradient in the Qinling Mountains of Central China: area-based versus mass-based expression of leaf traits
- Variations in stem radii of Larix principis-rupprechtii to environmental factors at two slope locations in the Liupan Mountains, northwest China
- A review of ecological mechanisms for management practices of protective forests
- Variation of basic density, calorific value and volumetric shrinkage within tree height and tree age of Ugandan grown Eucalyptus grandis wood