
MicroRNA-29a attenuates angiotensin-II induced-left ventricular remodeling by inhibiting collagen, TGF-β and SMAD2/3 expression
2020-03-05 03:03:08SiJinZHANGCuiJuanYUNJieLIUSiYuYAOYaoLIMiaoWANGChiWANGYongYiBAIHaoXUE
Journal of Geriatric Cardiology 2020年2期
Si-Jin ZHANG, Cui-Juan YUN, Jie LIU, Si-Yu YAO, Yao LI, Miao WANG, Chi WANG, Yong-Yi BAI, Hao XUE,#
1Department of Cardiology, Chinese People’s Liberation Army General Hospital, Beijing, China
2Department of Geriatric Cardiology, Chinese People’s Liberation Army General Hospital, Beijing, China
Abstract Background Left ventricular (LV) remodeling is the most common target organ damage in hypertension. Previously, our study found that plasma microRNA-29a (miR-29a) level was associated with the LV remodeling in hypertensive patients. However, the causal relationship between miR-29a and LV remodeling remains unknown. Thus, the aim of this study was to investigate the regulation mechanism of miR-29a in LV remodeling. Methods & Results Overexpression and knockdown miR-29a mice were generated by tail-intravenous injection of miR-29a-mimic and inhibitor lentivirus for one week respectively. Then the mice were subjected to angiotensin-II (AngII) induced LV remodeling by subcutaneous AngII capsule osmotic pumping into AngII for four weeks. AngII-induced LV remodeling mice as the model group (n = 9). Age-matched male SPF C57/BL6J mice (6-8 weeks old) were treated with the pumping of saline as a vehicle (n = 6). In vivo, overexpression miR-29a ameliorated AngII-induced LV remodeling, while knockdown miR-29a deteriorated LV remodeling. Simultaneously, we observed that overexpression miR-29a mice inhibited but knockdown miR-29a mice increased cardiac cross-sectional area, indicating that miR-29a has an antagonistic effect on cardiac hypertrophy. Further studies found that overexpression miR-29a inhibited the content of the LV collagen including collagen I and III. Moreover, the expression of transforming growth factor-β (TGF-β) and phosphorylated SMAD2/3 decreased with the down-regulation of collagen I and III in overexpression miR-29a mice. Conclusions Our finding indicates that overexpression miR-29a attenuates LV remodeling by inhibiting collagen deposition, TGF-β, and phosphorylated SMAD2/3 expression. Thus, intervention miR-29a may be a therapeutic target for attenuating LV remodeling.
J Geriatr Cardiol 2020; 17: 96-104. doi:10.11909/j.issn.1671-5411.2020.02.008
Keywords: Left ventricular remodeling; MicroRNA-29a; SMAD2/3; Transforming growth factor-β
1 Introduction
Left ventricular (LV) remodeling is the most common target organ damage in hypertension, which is also a critical cause of cardiovascular disease such as coronary heart disease, heart failure, arrhythmia, atrial fibrillation.[1]A previous study found that the prevalence of LV hypertrophy (LVH) is 42.7% in hypertensive patients among Han Chinese.[2]Hypertension is the main cause of LV remodeling, effective control blood pressure cannot completely prevent the occurrence of LV remodeling, suggesting other possible causes. Recent studies have found that epigenetics involved in the occurrence and development of ventricular remodeling.[3,4]
MicroRNAs (miRNAs) are endogenous non-coding small RNAs that regulate gene expression. Accumulating studies have found that miRNAs play important roles in regulating cardiovascular disease.[5-11]Several miRNAs are shown to be involved in cardiac remodeling, namely: miR-1, miR- 133, miR-26, miR-9, and miR-98 inhibit cardiac remodeling, and miR-143, miR-199a, miR-208, miR-23, miR-499, miR- 21 contribute to cardiac remodeling.[12-17]However, the molecular mechanisms of individual miRNAs remain unknown. A recent study found that microRNA-29a (miR-29a) prevented myocardial fibrosis after acute myocardial infarction by down-regulating the expression of extracellular matrix collagen.[18,19]In addition, the up-regulation of miR-29a was associated with cardiac hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy.[20]Our previous study also found circulating miR-29a is associated with the LV remodeling in hypertensive patients.[21]However, the regulatory mechanism of miR-29a in the remodeling process is still unclear.
In order to address this issue, we used overexpression and knockdown miR-29a mice model to investigate the regulation mechanism of miR-29a in LV remodeling.
2 Materials and methods
2.1 Animals
All the studies were performed in accordance with the relevant guidelines and regulations,[22]and approved by the Ethics Committee for Animal Study of Chinese PLA General Hospital. Healthy male SPF C57/BL6J mice (6-8 weeks old) were randomly assigned to four groups: (1) control group (pumping of saline, n = 6); (2) model group [pumping of angiotensin-II (AngII), n = 9]; (3) miR-29a-mimic group (n = 9) with injection of miR-29a-mimic lentivirus; and (4) miR-29a-knockdown (miR-29a-KD) group (n = 9) with injection of miR-29a-inhibitor lentivirus.
2.2 Generation of recombinant lentivirus
The short hairpin RNAs (shRNAs) containing mmu-mir- 29a (miR-29a-mimic, sequence: 5'-ACCCCTTAGAGGAT GACTGATTTCTTTTGGTGTTCAGAGTCAATAGAAT TTTCTAGCACCATCTGAAATCGGTTATAATGATTG GGGA-3') or a specific inhibitor of mmu-mir-29a (miR- 29a-KD, sequence: 5'-TAGCACCAGACTAAATCGGTT AAAGTAGCACCACAGTAAATCGGTTAGGTTAGCA CCACACAAAATCGGTTACAATAGCACCACAGAAA ATCGGTTAGTCTAGCACCAGTCAAAATCGGTTAAG GTAGCACCACTGAAAATCGGTTATACTAGCACCA GTGTAAATCGGTTATGATAGCACCAGTGAAAATC GGTTA-3') were cloned into the lentiviral vector pHS- AMR. shRNAs containing an unrelated sequence were used as negative controls, sequence: 5'-TAATTGTCAAA TCAGAGTGCTTGTTTTGGCCACTGACTGACAAGCA CTATTTGACAATTA-3'. Viral production was performed by using EpFectTMTransfection Reagent for the transfection of human embryonic kidney (HEK)-293 cells. Virus was concentrated by ultracentrifugation. Recipient cells were infected with 106viral transducing units per mL plus polybrene and stably transduced cells were selected in puromycin for ten days or sorted by Fluorescence Activated Cell Sorter (FACS).
2.3 Construct model
Capsule osmotic pump was purchased from AlZET Corporation (MODEL 2004), and AngII was purchased from Sigma Corporation of the United States. The animals underwent tail-intravenously injection of recombinant lentivirus (109titer/mL, 0.1 mL per mouse). Overexpression (n = 9) and knockdown (n = 9) miR-29a mice were generated by tail-intravenous injection of miR-29a-mimic and inhibitor lentivirus one week respectively. The control group and the model group were injected with normal saline intravenously. Besides the control group, the mice were subjected to AngII by subcutaneous AngII capsule osmotic pump (1400 ng/kg·min-1) for four weeks to induce LV remodeling.
2.4 Ultrasound assessment
At the end of the experiment, the mice were weighed and anesthetized intraperitoneally with 2.5% Avertin (0.018 mL/g). Transthoracic echocardiography was performed in all mice (Vevo 2100 imagine system, FuJIFILM Visualsonic Inc., Toronto, Canada) with a high transducer frequency probe (VisualSonics MS400, 18 MHz to 38 MHz). The LV dimensions, LV anterior wall (LVAW) thicknesses, and LV posterior wall (LVPW) thickness were averaged from more than three cardiac cycles at end-diastole and end-systole by the M-mode measurements.
2.5 Tissue preparation
The mice thoracic cavity were immediately opened. The hearts were removed and perfused with saline under physiological pressure until the liver turned white. The cardiac tissue slices (3 mm thick) were cut in the mid-ventricle, fixed in 4% paraformaldehyde for 24 hours, flushed by phosphate buffered saline (PBS), dehydrated in ascending series of ethanol and embedded with paraffin. The remaining LV slices were quick-frozen in liquid nitrogen and stored under -80 °C.
2.6 Histological analysis
2.6.1 Masson and Sirius red staining
Paraffin-embedded tissue sections were stained with Masson and Sirius red staining separately. Masson staining was performed as follows: after being dehydrated in gradient ethanol, the sections were immersed into the Alcian Blue staining solution, acid alcohol, ponceau, and phosphomolybdic acid solution in sequence to stain, and the change of cardiac structure, myocardial cells and cardiac fibroblasts were observed with a microscope (200 ×). Sirius red staining was performed by incubating sections in Sirius red solution, washing in acidified water and then dehydrating in ethanol to observe the expression of the LV myocardial collagen. The cardiac cross-sectional area (mm2) and the LV collagen volume were measured through IPP 6.0.
2.6.2 Immunofluorescent detection
Paraffin-embedded sections were dewaxed, enclosed for 12 min in 3% H2O2and then flushed three times in PBS, repaired with microwave energy combined with Ethylenediaminetetraacetic Acid and flushed three times in PBS again. Subsequently, the sections were enclosed in fetal bovine serum protein solution for one hour at room temperature, after that, dropwise added first antibody of collagen I and III (1:50, Abcam and Proteintech), incubated for overnight in 4 °C. The next day, the sections were flushed three times in PBS firstly, and then dropwise added the goat secondary antibody labeled with fluorescence, incubated for one hour in 37 °C and finally stored in 4 °C without light. The expression of myocardial tissue collagen I and III was observed with a fluorescence microscope (400 ×) and the collagen area was measured by partial photography and IPP 6.0 software.
2.6.3 Western blotting analysis
The frozen LV tissue of the mice was added the tissue Lysis Buffer, then ground and centrifuged to get supernatant. The isovolumetric 2 × gel-loading buffer was added to the supernatant after protein excretion. Subsequently, the supernatant boiled for 10 min. The proteins were loaded onto 8% Sodium Dodecyl Sulfate polyacrylamide gel for separation, and transferred to nitrocellulose membrane, then marked the target protein band after the ponceau staining. The first antibody was enclosed for one min in 5% skimmed milk and then incubated for one min at room temperature, incubated overnight at 4 °C. The antibodies were then stripped by washing the membrane 3 × 10 min with 0.2% TPBS (Triton X-100/PBS). The secondary antibody was incubated for one hour at room temperature. The membrane was then rinsed 4 × 10 min in 0.2% TPBS. The primary proteins used were the first antibody of transforming growth factor-β (TGF- β) (Cell Signaling Technology, 1:1000), p-SMAD2/3 (Cell Signaling Technology, 1:1000), Collagen-I (Abcam, 1:500), Collagen-III (Proteintech, 1:500), β-actin (Abcam, 1:4000), and the secondary antibody of rabbit anti-goat IgG (1:2000), goat anti-mouse IgG (1:5000), goat anti-rabbit IgG (1:2000).
2.7 Statistical analysis
The experimental data analysis was performed by using SPSS 20.0. All values were presented as mean ± S.E. Statistical differences among the groups were performed by One-Way ANOVA. A P-value < 0.05 was considered as statistically significant.
3 Results
3.1 Overexpression of miR-29a inhibits LVH in vivo
To investigate the effect of miR-29a on LVH, we constructed overexpression and knockdown miR-29a mice. We found that ultrasound parameters of LVH were higher in the model group than those in the control group, indicating that the construction of the model was successful. And, compared with the model group, miR-29a overexpression obviously reduced the thickness of the LVAW at the end-diastolic and the LVPW at the end-systolic (as shown in Figure 1Ab-c and Figure 1Ba-d). Conversely, the miR-29a-KD increased the thickness of LVAW and LVPW. These results indicated that overexpression of miR-29a ameliorated LV remodeling induced by AngII, while knockdown miR-29a deteriorated left ventricular remodeling in vivo.
3.2 miR-29a inhibits fibroblast proliferation
We observed the histological changes in the heart. Masson staining showed that the cardiac cross-sectional area was significantly larger in the model group than that in the control group (Figure 2A). The cardiac cross-sectional area was reduced in miR-29a-mimic group but increased in the miR-29a-KD group (Figure 2Ca). These results indicated that miR-29a attenuated cardiac hypertrophy. The ratio of the collagen area in the LV area by Sirius Red Staining (Collagen-specific Dyeing) showed that compared with the model group, miR-29a overexpression obviously inhibited the content of collagen, suggesting that it inhibited fibroblast proliferation. However, we did not find that miR-29a- KD group had a significant effect on the ratio of the collagen area in the LV area (Figure 2Cb). These results indicated miR-29a improves the myocardial remodeling by inhibiting fibroblast proliferation.
3.3 miR-29a suppresses the secretion of collagen I and III
Immunofluorescent detection showed that compared with the model group, miR-29a overexpression significantly inhibited the levels of collagen I and III (Figure 3A & B); However, the levels of collagen I and III were no significant difference in the miR-29a-KD group. The result is consistent with the effect of miR-29a-mimic and miR-29a-KD on the ratio of the collagen area in the LV area (Figure 2), suggesting that miR-29a overexpression can dramatically inhibit secreting collagen I and III.
3.4 miR-29a decreases the expression of TGF-β and SMAD2/3

Figure 1. Overexpression of miR-29a inhibits left ventricular remodeling by using ultrasound assessment.
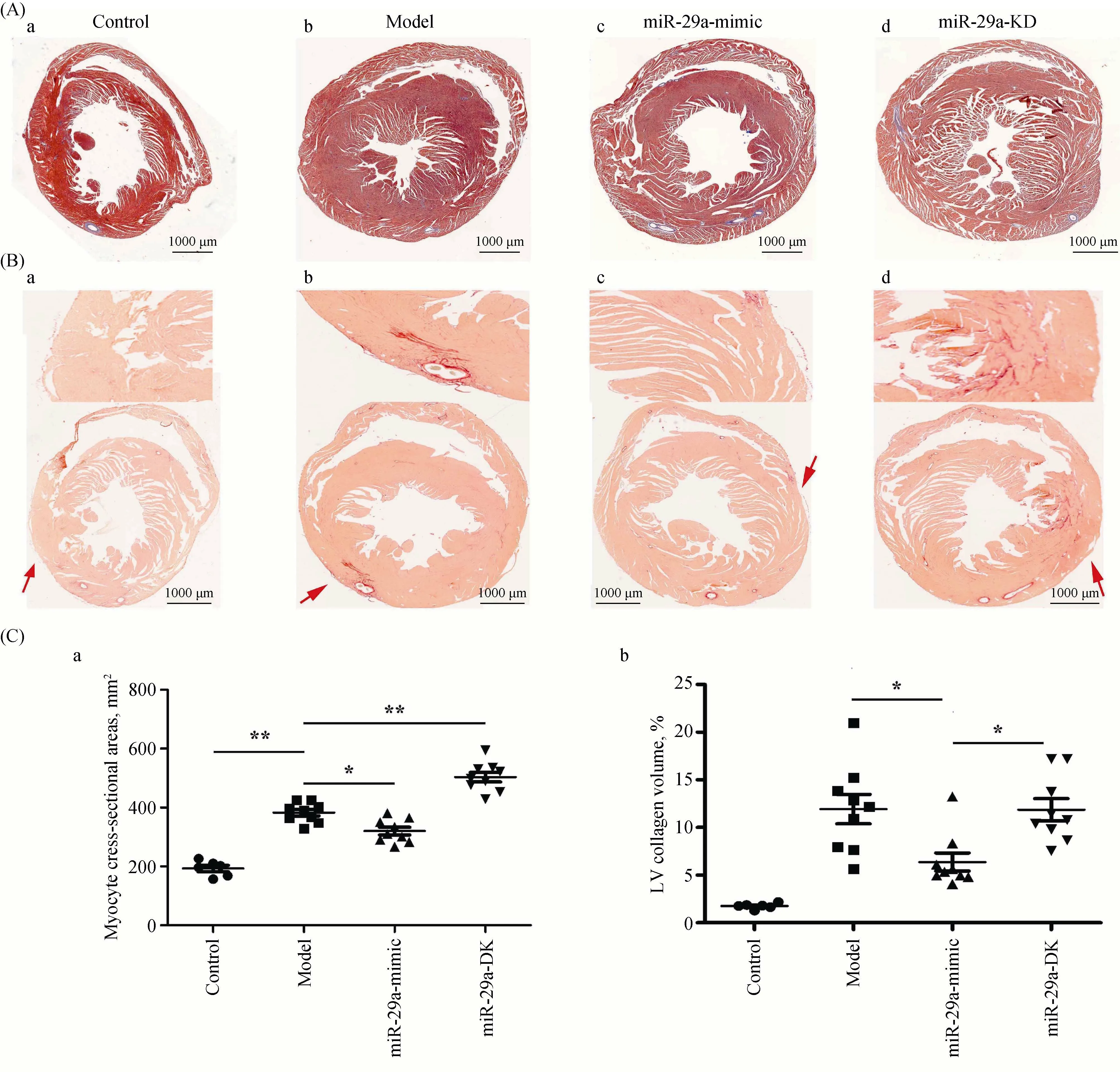
Figure 2. Overexpression of miR-29a inhibits fibroblast proliferation.
The TGF-β and SMAD2/3 pathways are classic ways for collagen activation and secretion of myocardial fibroblasts. Our results suggested miR-29a overexpression protected from LV remodeling (Figures 1-3). Therefore, we further investigated the cardiac expression of TGF-β and SMAD2/3 in the model group and miR-29a-mimic group by using Western blot. Compared with the model group, miR-29a overexpression significantly inhibited the expression of TGF-β and phosphorylated SMAD2/3 (activated form) in the heart, accompanied by a significant decrease in the expressing quantity of collagen I and III (Figure 4A & B). However, there was no obvious difference in the cardiac expression of TGF-β and SMAD2/3 between miR-29a-KD group and model group (Figure 4C & D). These results indicated that overexpression miR-29a can reduce cardiomyocyte proliferation to ameliorated LV remodeling by inhibiting the content of LV collagen including collagen I and III, accompanied by decreasing TGF-β and phosphorylated SMAD2/3.
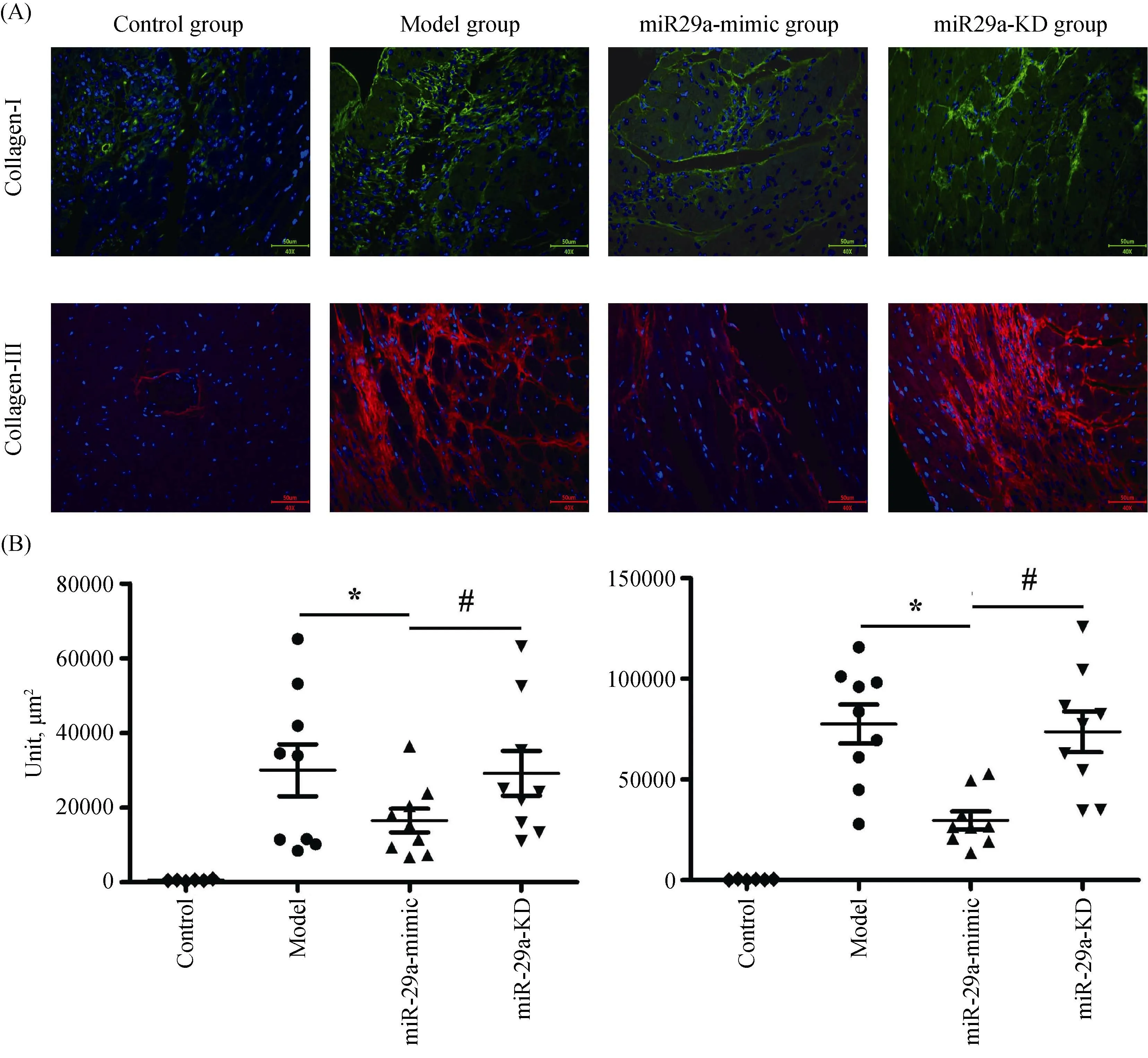
Figure 3. Inhibition of miR-29a on cardiac collagen.
4 Discussion
This is the first vivo study to investigate the mechanism of miR-29a in regulating LV remodeling. We found that cardiac overexpression of miR-29a attenuated cardiac remodeling, accompanied by collagen I, collagen III, TGF-β and phosphorylated SMAD2/3 expression decreasing.
Cardiac remodeling is a chronic pathological process accompanied by cardiac hypertrophy and myocardial fibrosis. Previous studies suggested that miR-29 protected the heart from pathological hypertrophy and myocardial fibrosis.[19,23]In patients with hypertrophic cardiomyopathy, circulating miR-29a was identified to be a biomarker for both hypertrophy and fibrosis.[20]The consistent results have also shown that miR-29a is positively correlated with LVH in hypertension patients.[24]In addition, experimental cardiac remodeling and reverse remodeling animal model con- firmed that miR-29a involved in the cardiac reverse remodeling process.[25]Our study adds new evidence for the protective role of miR-29a in overexpression miR-29a mice and knockdown miR-29a mice subjected to AngII induced LV remodeling. We observed that overexpression miR-29a inhibited but knockdown miR-29a aggravated AngII-induced LV remodeling in vivo and pathological changes of cardiac remodeling (including cardiac cross-sectional area, and cardiac fibrosis), which also indicated the protective role of miR-29a in cardiac remodeling.
Accumulation of extracellular matrix molecules is one of the main pathological features of LV remodeling.[26,27]Previous research showed that miR-29a was produced mostly by fibroblasts, and its family members regulated fibrosis by modulating the expression of collagen and other extracellular matrix genes, such as TGF-β, vascular endothelial growth
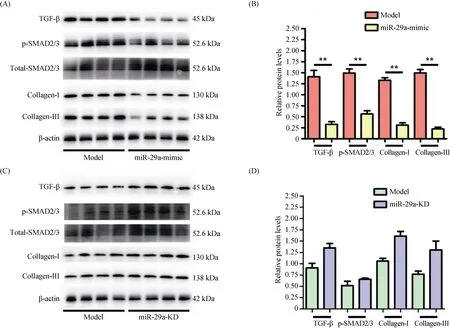
Figure 4. Inhibition of miR-29a on the expression of Collagen I, Collagen III, TGF-β and SMAD2/3.
factor-A, fibrillins, elastin, and myeloid cell leukemia 1 gene.[18,28-30]In our study, miR-29a overexpression obviously decreased the content of LV collagen including collagen I and III, which indicated that it inhibited fibroblast proliferation. This finding is consistent with the results reported by Melo, et al.,[19]which shows swimming training- induced up-regulation of the expression of miR-29 is related to repression of collagen (collagen I and III) gene expression in myocardial-infarcted mice. In addition, by using bioinformatics websites (Target Scan Human), we also predicted that collagen I and III might be a target of miR-29a, which may participate in the regulation of cardiac hypertrophy. However, we did not find significant up-regulation of collagen I and III expression in knockdown miR-29a mice. Several reasons might explain the conflicting results. Firstly, exogenous delivery of the miR-29a inhibitor might not completely knock down cardiac-specific miR-29a expression. Secondly, miR-29a may not be the sole determinant of collagen expression, and other factors may also involve in it.
Another finding of our study was that the expression of TGF-β and phosphorylated SMAD2/3 was decreased accompanied by the down-regulation of collagen I and III in miR-29a-mimic group, whereas knockdown of miR-29a increased the expression. The results indicated that TGF-β and SMAD2/3 may involve in the regulation mechanism of miR-29a in LV remodeling. It has been well known that TGF-β is identified as a regulator of cardiac fibrosis,[31,32]which can promote collagen protein synthesis. TGF-β and SMAD signaling pathway was considered to be one of the most classic ways of mediating fibroblasts activation and collagen secretion.[33]Moreover, other studies indicated that TGF-β repressed miR-29 expression.[18,34]However, overexpression miR-29a can also inhibit TGF-β and SMAD2/3 expression. These data demonstrate that TGF-β and SMAD may be upstream signaling pathway molecules of miR-29a target gene collagen in regulating cardiac hypertrophy.
In the present study, we found that miR-29a attenuated cardiac remodeling accompanied by lowing collagen, TGF-β and phosphorylated SMAD2/3 expression. However, the signaling pathway of miR-29a targeted collagen in the regulation of LV remodeling could not be fully explained. Further studies are needed to reveal the complete mechanism of miR-29a expression and its association with the signaling pathway of target gene collagen.
In summary, our study showed that overexpression miR- 29a contributed to attenuate AngII-induced cardiac remodeling by down-regulating collagen, TGF-β and phosphorylated SMAD2/3 expression. Thus, miR-29a may be a potential therapeutic target for inhibiting hypertensive LV remodeling.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No. 81570383). All authors had no conflicts of interest to disclose.
 Journal of Geriatric Cardiology2020年2期
Journal of Geriatric Cardiology2020年2期
- Journal of Geriatric Cardiology的其它文章
- Diagnostic chest X-ray in atrial septal defects
- Short-term efficacy of unibody single-branched stent in the treatment of lesions involving the left subclavian artery: two-year follow-up outcomes
- Is it better to choose immediate dialysis treatment for renal transplant patients after PCI?
- A novel treatment of refractory coronary embolism: thrombus aspiration catheter-assisted twisting wire technique
- Beneficial effects of moderate to vigorous physical activity on cardiovascular disease among Chinese adults
- Atrial fibrillation among Russian men and women aged 55 years and older: prevalence, mortality, and associations with biomarkers in a population-based study