
Paleo-polyploidization in Lycophytes
2020-03-04 09:21:28JinpengWangJigaoYuPengchuanSunChaoLiXiaomingSongTianyuLeiYuxianLiJiaqingYuanSangrongSunHonglingDingXueqianDuanShaoqiShenYanshuangShenJingLiFanboMengYangqinXieJianyuWangYueHouJinZhangXianchunZhangXiuQingLiAndrewP
Jinpeng Wang,Jigao Yu,Pengchuan Sun,Chao Li,Xiaoming Song,2,Tianyu Lei,Yuxian Li,Jiaqing Yuan,Sangrong Sun,Hongling Ding,Xueqian Duan,Shaoqi Shen,Yanshuang Shen,Jing Li,Fanbo Meng,Yangqin Xie,Jianyu Wang,Yue Hou,Jin Zhang,Xianchun Zhang,Xiu-Qing Li,Andrew H.Paterson,Xiyin Wang,2,
1 Center for Computational Biology and Genomics,and School of Life Sciences,North China University of Science and Technology,Tangshan 063200,China
2 National Key Laboratory for North China Crop Improvement and Regulation,Agriculture University of Hebei,Baoding 071001,China
3 State Key Laboratory of Systematic and Evolutionary Botany,Institute of Botany,Chinese Academy of Science,Beijing 100093,China
4 Fredericton Research and Development Centre,Agriculture and Agri-Food Canada,Fredericton,New Brunswick,E3B 4Z7,Canada
5 Plant Genome Mapping Laboratory,University of Athens,Athens,GA 30602,USA
6 Department of Plant Biology,University of Georgia,Athens,GA 30602,USA
7 Department of Crop and Soil Science,University of Georgia,Athens,GA 30602,USA
8 Department of Genetics,University of Georgia,Athens,GA 30602,USA
Abstract Lycophytes and seed plants constitute the typical vascular plants.Lycophytes have been thought to have no paleo-polyploidization although the event is known to be critical for the fast expansion of seed plants.Here,genomic analyses including the homologous gene dot plot analysis detected multiple paleo-polyploidization events,with one occurring approximately 13-15 million years ago (MYA) and another about 125-142 MYA,during the evolution of the genome of Selaginella moellendorffii,a model lycophyte.In addition,comparative analysis of reconstructed ancestral genomes of lycophytes and angiosperms suggested that lycophytes were affected by more paleopolyploidization events than seed plants.Results from the present genomic analyses indicate that paleo-polyploidization has contributed to the successful establishment of both lineages—lycophytes and seed plants—of vascular plants.
KEYWORDS Vascular plant;Lycophytes;Genome;Polyploidy;Evolution
Introduction
Extant vascular plants can be divided into two major types,the euphyllophytes (ferns and seed plants) and the lycophytes,which diverged as early as 410 million years ago (MYA) [1].Selaginella moellendorffii,as a model lycophyte,has a dominant and complex sporophyte generation and vascular tissues with lignified cell types.The deciphered genome ofS.moellendorffiiis approximately 212.6 Mb,including two haplotypes of approximately 106 Mb [1].Recently,the even smaller genome ofS.lepidophylla,approximately 109 Mb,was deciphered [2].
Recursive polyploidizations or whole-genome duplications have been proposed as a key evolutionary driving force of seed plants,contributing to their divergence and rapid expansion[3,4].Different from seed plants,genomic analyses with various approaches including a discontiguous MegaBLAST did not find evidence of paleo-polyploidization in bothS.moellendorffiiandS.lepidophyllagenomes [1,5].
Seed plant genomes can be quite complex,at least partially due to recursive polyploidization and repetitive sequence accumulation.It is often difficult to perform a comprehensive analysis to understand their genome structure and evolution.In several instances,ancestral polyploidization was omitted from genome analysis,resulting in problematic interpretation of the structure,evolution,and/or functional innovation of whole genomes and key gene families[6-9].Recently,we developed a pipeline [10],including the generation of dot plot chart of homologous genes[11],to facilitate the analysis of complex genomes,especially those of plants.Analysis of the cucurbit genomes using this pipeline revealed an overlooked paleotetraploidization event,occurring~100 MYA,in the common ancestor of Cucurbiticeae plants,which possibly contributed to the establishment and fast divergence of the whole family[10].
Here,we used our pipeline [10],including the homologous gene dot plot analysis [11],to analyze bothSelaginellagenomes and other related plant genomes [12].We detected multiple polyploidization events in lycophytes.
Results
Gene colinearity
Using a gene-colinearity-based approach implemented in ColinearScan,with maximal gap between neighboring colinear genes of 50 genes (consistent with prior studies of many plants),we inferred 302 syntenic blocks in theS.moellendorffiigenome(Tables S1 and S2).These blocks involved 2632 colinear gene pairs,and covered 87.01% (19,391/22,285) of all genes and 88.24% (19,391/21,975) of assembled genome sequences (Figure 1A).Use of a smaller gap size (25 genes)does not qualitatively change this result (65.92%genome coverage).The assembly had 192 scaffolds (of at least five genes),of which at least 75.52% (145/192) were covered by syntenic blocks with five or more colinear genes (more information about blocks can be found in Table S3).
The syntenic blocks in theS.moellendorffiigenome were mapped onto the chromosomes and produced coverage as deep as 10.Approximately 19.24% of genome regions were covered with a depth of 4 or more,and 17.55%,25.36%,27.06%,and 10.79% of regions were covered with a depth of 3,2,1,and 0,respectively(Figure S1 and Table S4).Correspondence of colinear genes between multiple duplicated regions could often be found,showing probable recursive gene duplications (Figure 1B).
Evidence of recursive polyploidization events
Sequence divergence of colinear genes in theS.moellendorffiigenome clearly showed a non-random distribution consistent with the occurrence of polyploidization.Both synonymous nucleotide substitutions per synonymous site (Ks) and nucleotide diversity for the fourfold synonymous thirdcodon transversion position (4Dtv) displayed clear bimodal distributions (Figure 2,Figures S2 and S3).ForKs,the two peaks were located at 0.12 and 1.2,respectively(Figure 2).Based on theKs distribution,we divided the colinear genes into two groups (Figure 1A).Supposing 6.0-7.0 × 10-9synonymous substitutions per site per year,borrowed from angiosperms [3,13,14],the two peaks were reposited at 0.12 and 1.15,respectively,suggesting large-scale genomic duplication events to have occurred~13-15 MYA(named ξ or Xi) and~125-142 MYA (named η or Eta).The use of separate distributions rather than the merged distribution allowed elimination of interactive effects during statistical analysis.The younger group covered 76.65%(16,844/21,975 genes) of the genome and 70.31% (135/192)of total scaffolds (related to all chromosomes).This result indicated that the event ξ was a polyploidization,with aKs peak clearly distinct from any residual heterozygosity ofS.moellendorffii(DNA similarity~98.5% [1],corresponding toKs~0.056).
By merging the colinear gene blocks generated from the polyploidization event 13-15 MYA,we inferred that the ancestral genome before this event consisted of at least 120 syntenic blocks,which included 936 colinear genes,and covered 64.79%(8434/13,017)of the ancestral genome.We also discovered the existence of a more ancient whole-genome doubling event (the 125-142 η event),because self-comparison of these 120 blocks showed further colinearity that is extremely improbable to occur by chance.
Comparison between S.lepidophylla and angiosperms
Furthermore,we checked whether the two ancient polyploidization events detected in theS.moellendorffiigenome was shared withS.lepidophyllaand seed plants.Though twoSelaginellagenomes shared obvious gene colinearity(Tables S1 and S2),an analysis of theS.lepidophyllagenome did not find evidence of polyploidization.The two lycophytes have 1121 colinear blocks with length ≥4 colinear genes,including 11,087 colinear genes in total.InS.lepidophylla,110 blocks including 538 colinear genes were found,much fewer than inS.moellendorffii.By characterizingKs between colinear genes,we foundKs peaks of putative orthologous genes at 1.35,1.57,and 1.60 forS.moellendorffii vs.S.lepidophylla,Amborella trichopoda(a model angiosperm),andVitis vinifera(grape),respectively (Figure 2).This result suggested that the two inferred polyploidization events in theS.moellendorffiilineage were absent inS.lepidophyllaand in angiosperms (Figure 3).
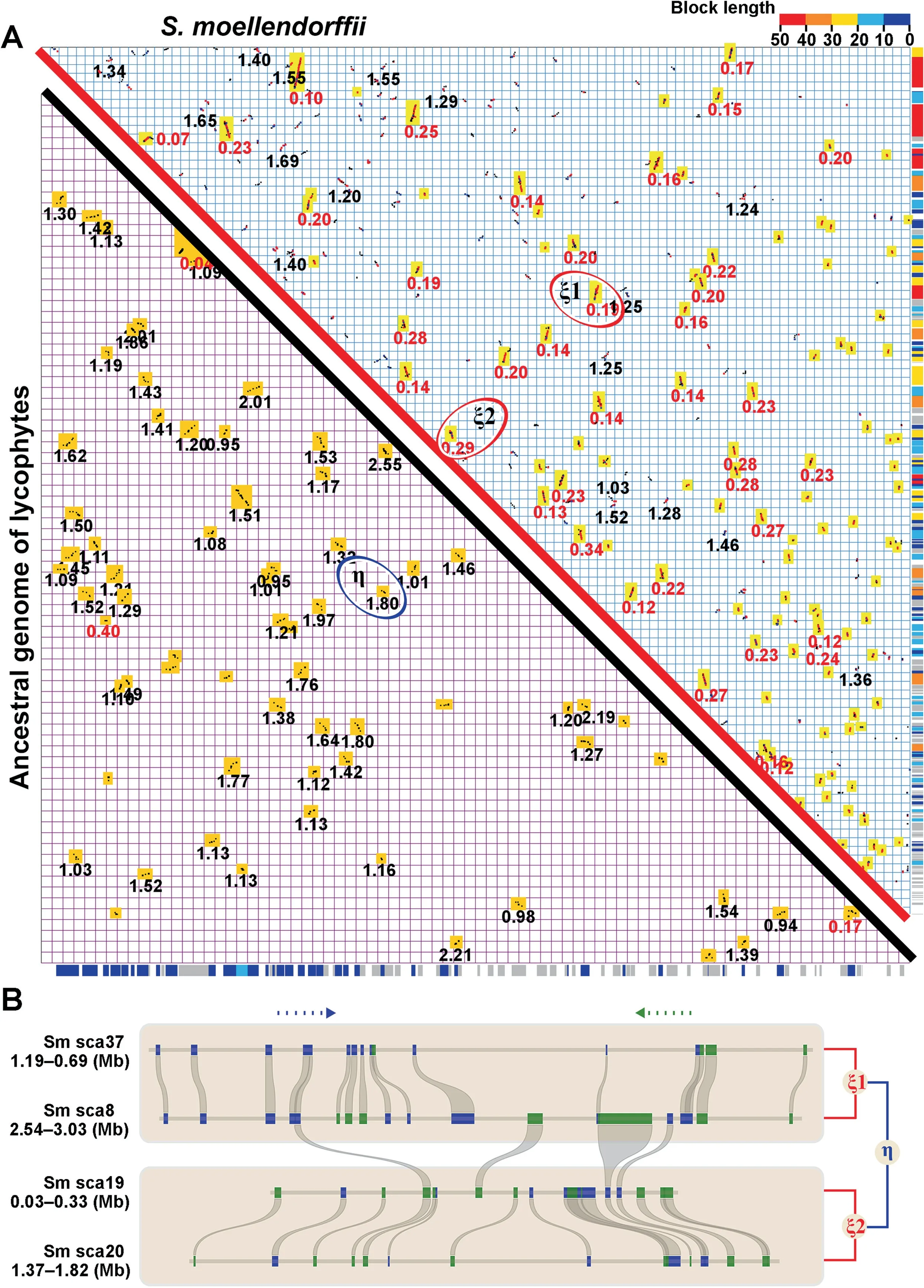
Figure 1 Homologous gene dot plot of Selaginella moellendorffii and alignment of genomic regions with colinear genes
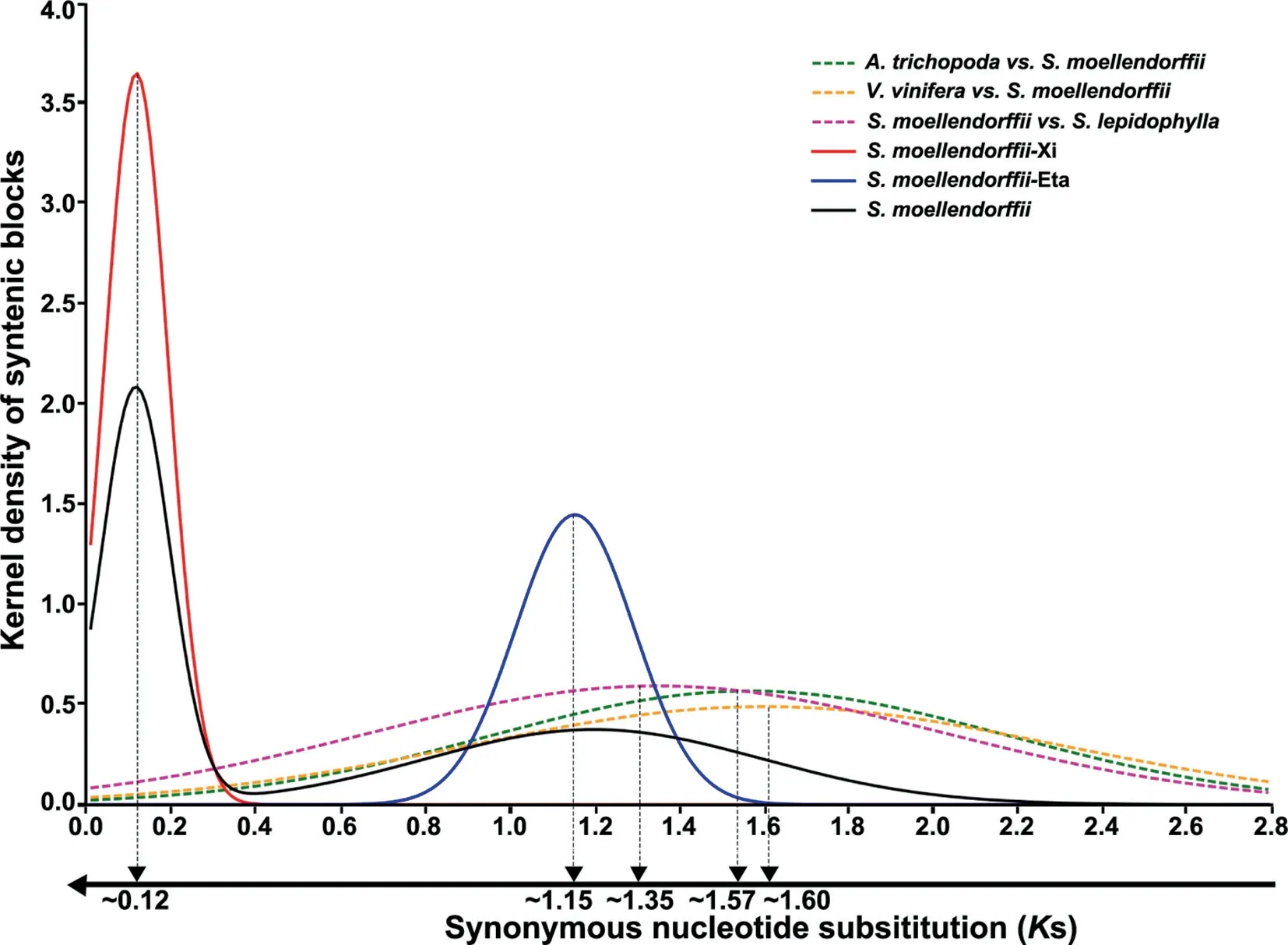
Figure 2 Distribution of Ks between colinear genes
Existence of likely more paleo-polyploidization events
An appreciable fraction ofS.moellendorffiigenomic regions were covered at a depth >4 by colinear blocks,suggesting more ancient polyploidization.Therefore,we performed a deeper search of gene synteny betweenS.moellendorffiiand the two referenced angiosperm genomes.TheAmborellagenome,assembled into scaffolds,has a relatively simple structure and avoided a genome doubling after its split from other angiosperms,despite the existence of evidence of a shared ancient polyploidization[15](Figure 3).The grape genome,well assembled into pseudochromosomes,was also used to reconstruct the ancestral genome before the major-eudicot-common hexaploidy.We inferred syntenic blocks betweenS.moellendorffiiand each of the referenced genomes and mapped the blocks onto each of the genomes involved(Figures S4 and S5).Totals of 14.18% and 9.73% of theS.moellendorffiiandAmborellagenomes,respectively,were covered by colinear blocks to depth of 4 or more(Table S5).Likewise,21.34%and 13.86%of theS.moellendorffiiand grape genomes,respectively,were covered by colinear blocks to depth of 4 or more(Table S6).These findings suggested additional more ancient polyploidization events during the evolution of vascular plants.
We then explored the coverage depth of homologous regions in the reconstructed pre-ξ ancestral genome of lycophytes (ALG,11,509 genes) and ancestral genome of angiosperms (AAG,1686 genes) inferred on the basis of the grape genome.The ALG involved genes from the 34 largestS.moellendorffiiscaffolds.At a gap size of 50 genes using ColinearScan,significant colinear blocks (i.e.,with at least four colinear genes)resulted in homologous coverage depth as deep as 7 and 18 in the AAG and ALG,respectively (Figure 4).Indeed,78.28% and 7.49% of the AAG and ALG,respectively,were covered to depth of 4 or more (Table S7).To be careful,we tried different maximal gap sizes in each compared lineage(Table S7) and obtained similar conclusions (Figure 3).
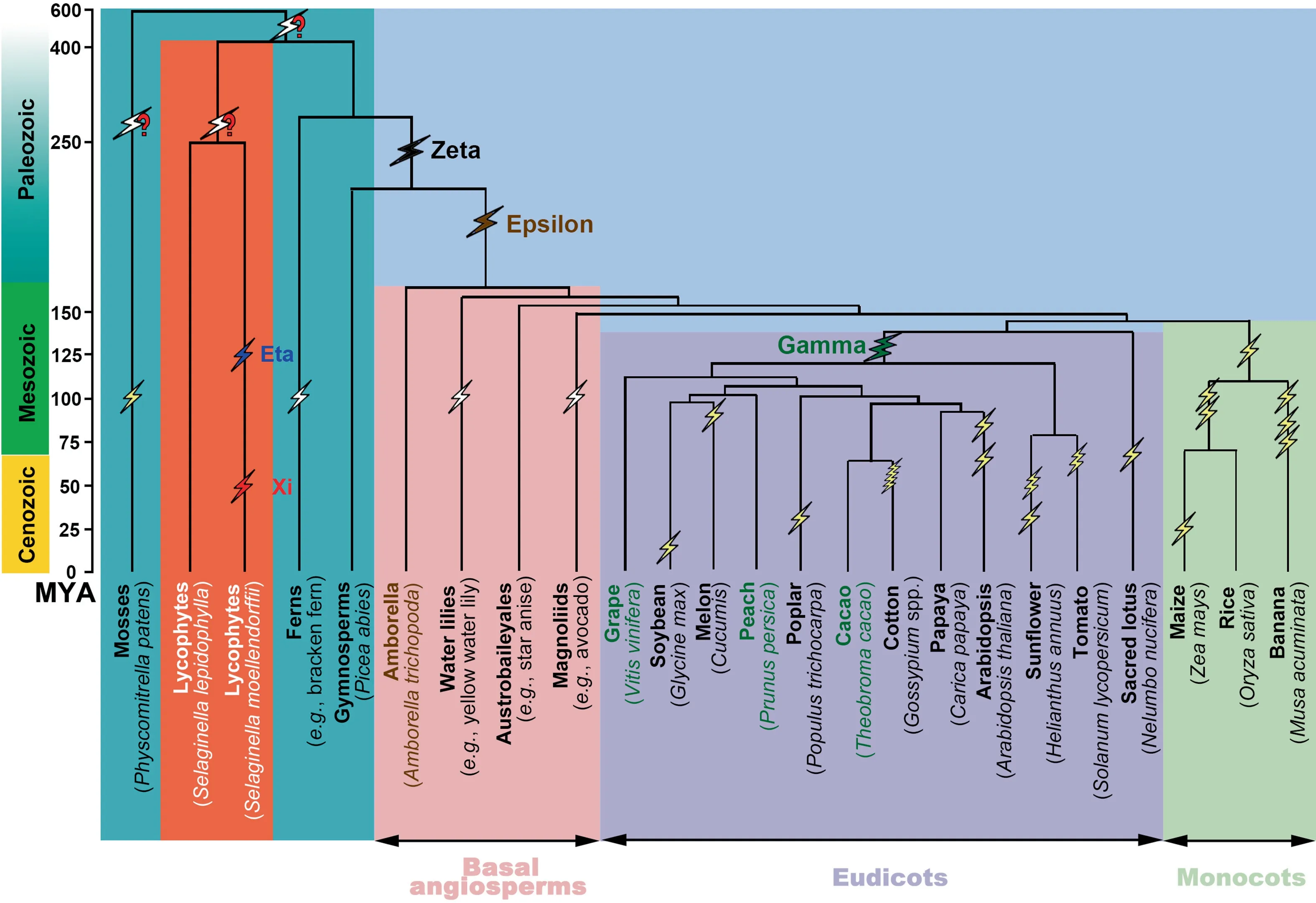
Figure 3 Plant phylogeny and inferred ancient polyploidies
In thatS.lepidophyllawas not affected by polyploidization after its divergence withS.moellendorffii,it could provide insight into polyploidization(s) older than their split from angiosperms.Therefore,we managed to useS.lepidophyllagenomic DNA,though not as well assembled as that ofS.moellendorffii,to infer gene colinearity.TheS.lepidophyllacontigs were sorted as to their best matchedS.moellendorffiiscaffolds.We found appreciable amount of colinearity within the genome itself,and betweenS.lepidophyllaand AAG (Figure S6 and Table S1).Regarding coverage depth,68.55% and 20.74% of the AAG andS.lepidophylla,respectively,were covered to depth of 4 or more (Table S8).At least 40% of the inferred colinear blocks terminate at the ends of assembled scaffolds,presumably broken by incomplete assembly rather than by lack of colinearity (Figure S7).
Since lycophytes and seed plants are thought to have diverged 410 MYA[1],and the twoSelaginellaplants(S.moellendorffiiandS.lepidophylla) diverged approximately 250 MYA [2],the present findings of at least two paleopolyploidization events in less than 150 MYA suggest that there may have been additional paleo-polyploidizations in each lineage of lycophytes and seed plants,separately.Moreover,greater coverage mapped onto the AAG means that lycophytes might have experienced more polyploidizations than seed plants.
We have to note that a more precise determination of the numbers and dates of these ancient polyploidization events is beyond the resolution ofKs.Other sequence divergence analysis approaches using likely more taxa of basal vascular plants will be required.
Discussion
Polyploidization or whole-genome duplication was inferred to have contributed to the early divergence of seed plants according to phylogenetic tree reconstruction [4].By dissecting features ofKs distribution with transcriptome data,horsetail,a basal land plant,was found to be an ancient polyploid[16].A large-scale cytogenetic and phylogenetic analysis indicated that ferns might have been more frequently affected by polyploidization [17].Together with many other publications proposing ancient polyploidization [18-21],these works each shed light on the distant past of plant evolution.However,although the firstSelaginellagenome has been available for years,genome-scale evidence of ancient whole-genome duplication in the lycophytes remains elusive.Here,using the sophisticated pipeline that we developed for deciphering complex genomes,we provided clear and solid evidence that two polyploidization events have contributed to the evolution of the lycophyteS.moellendorffiiafter its split fromS.lepidophylla,and several older polyploidization events likely shared also bySelaginellaplants have occurred in the early days of vascular plants.
Polyploidizations were proposed to be the likely answer to the mystery about the abrupt origin and fast divergence of seed and flowering plants on Earth raised by Charles Darwin [20].The present finding that lycophytes were also recursively affected by polyploidizations may benefit further evaluation how these events contributed to the expansion of different lineages of vascular plants.
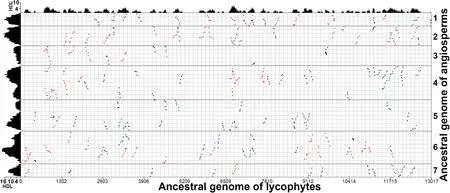
Figure 4 Inferred colinear genes between ancestral genome of lycophytes (ALG) and ancestral genome of angiosperms (AAG)
The availability of whole-genome sequences provides evidence of ancient polyploidy.However,it is technically challenging to deconvolute these ancient large-scale events [6-9].The MegaBLAST analysis of theSelaginellagenome overlooked these large-scale evolutionary events [1],possibly due to the complicated nature of plant genomes and/or drawbacks in the methodology adopted [10].To decipher the complex genomes,genome dot plot often makes a good illustration of gene synteny/colinearity.Divergence of inferred colinear genes reveals non-random patterns that distinguish subsets with different ages.The exploration of colinear genes can provide insight into more ancient events,as proposed,and can be successfully used to decipher recursive polyploidizations during the evolution of eudicots based on the small genome ofArabidopsis[11].Methodology that lacks inference of gene colinearity or use of homologous gene dot plots,e.g.,based largely on inferences ofKs distribution or phylogenetic tree topology,could easily overlook evidence of paleo-polyploidization.EvenKs distributions that give the appearance of polyploidization could be caused by genes duplicated by other events,such as waves of retrotransposition.The combination of temporal (Ks) and positional (colinearity) evidence can provide for falsification of alternative hypotheses,to uncover ancient,especially recursive polyploidizations in extant complex genomes.Future research may help to understand how these polyploidization events have contributed to biological and genetic innovations during evolution.
Conclusion
A major lineage of land plants,lycophytes,was thought to have avoided paleo-polyploidization.Here,using a sophisticated comparative genomics approach,we inferred that the genome of the first sequenced lycophyteS.moellendorffiiwas affected by at least two rounds of paleo-polyploidization events;and more analyses with another lycophyte and seed plants suggested more large-scale genome duplications during the evolution of land plants.
Materials and methods
Materials
Genome sequences and annotations were downloaded from the Joint Genome Institute (JGI,https://phytozome.jgi.doe.-gov/pz/portal.html;S.moellendorffii,S.lepidophylla,A.trichopoda,andV.vinifera L.).
Gene colinearity
The pipeline to decipher complex genomes was followed as described previously [10].Homologous gene dot plots within a genome or between genomes were produced by using MCSCAN toolkits [22],of which the corresponding author directly contributed to the development.Colinear genes were inferred by using ColinearScan [23],a statistically wellsupported software in pairwise homology.ColinearScan implements a modified dynamic programming algorithm and recursively searches the remaining longest path with colinear gene pairs as nodes.The maximal gap between genes involved in colinearity along a chromosome was evaluated computationally,and each colinear block inferred was then statistically assessed of their significance.Here,the maximal gap was set as 50 genes apart,and repetitive genes with >30 copies were removed from the present analysis,as adopted in many previous studies[6,24-27].Evolutionary divergence between homologous genes was estimated as described previously [24,26].
Analysis of Ks
The synonymous nucleotide substitutions (Ks) between duplicated genes in colinearity resulted from each of considered polyploidization and divergence events were analyzed.Ks values were estimated using Nei-Gojobori approach by implementing the program codeml in the ‘‘phylogenetic analysis by maximum likelihood”software PAML [28,29].Gene CDS alignment was performed using ClustalW with default parameters [30].
Construction of ancestral genomes
Construction of ancestral genomes was also based on our previously detailed methods [10,11].For clarification,here,we reconstructed the ancestral genome of lycophytes (ALG)before the polyploidization ξ,and the ancestral genome of angiosperms (AAG).The ALG was reconstructed by utilization of one copy of the colinear genes produced by the recent polyploidy,in that the colinear genes probably preserved their ancestral gene locations (Figure S8A).In a similar approach,the AAG genome was reconstructed by inferring ancestral genes using grape colinear genes produced by the majoreudicot-common hexaploidy (previously named Υ) (Figure S8B).We did not infer AAG using a grape-Amborellaor grape-rice comparison due to incomplete assembly of theAmborellagenome and severe genomic fractionation after multiple polyploidies in monocot lineages [31].
CRediT author statement
Jinpeng Wang:Writing -original draft,Supervision,Methodology,Software.Jigao Yu:Software,Formal analysis,Data curation,Visualization.Pengchuan Sun:Software,Formal analysis,Visualization.Chao Li:Software,Formal analysis.Xiaoming Song:Software,Formal analysis,Data curation.Tianyu Lei:Software,Formal analysis.Yuxian Li:Software,Formal analysis,Data curation.Jiaqing Yuan:Software,Formal analysis,Data curation.Sangrong Sun:Formal analysis,Data curation.Hongling Ding:Formal analysis,Data curation.Xueqian Duan:Formal analysis,Data curation.Shaoqi Shen:Formal analysis,Data curation.Yanshuang Shen:Formal analysis,Data curation.Jing Li:Formal analysis,Data curation.Fanbo Meng:Investigation,Data curation.Yangqin Xie:Investigation,Data curation.Jianyu Wang:Investigation,Data curation.Yue Hou:Data curation.Jin Zhang:Data curation.Xianchun Zhang:Validation.Xiu-Qing Li:Writing -review&editing.Andrew H.Paterson:Writing-review&editing.Xiyin Wang:Conceptualization,Writing -original draft,Writing -review &editing,Supervision.All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
We appreciate financial support from the Ministry of Science and Technology of the People’s Republic of China (Grant No.2016YFD0101001),the China National Science Foundation (Grant Nos.31371282 to XW,31510333 to JW,and 31661143009 to XW),the Natural Science Foundation of Hebei Province (Grant No.C2015209069 to JW),and Tangshan Key Laboratory Project to XW.We thank the helpful discussion with researchers at iGeno Co.Ltd,China.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2020.10.002.
ORCID
0000-0002-2253-7752 (Jinpeng Wang)
0000-0001-9156-2532 (Jigao Yu)
0000-0002-8999-4894 (Pengchuan Sun)
0000-0002-9607-6899 (Chao Li)
0000-0003-0084-3668 (Xiaoming Song)
0000-0001-9151-213X (Tianyu Lei)
0000-0002-2075-6724 (Yuxian Li)
0000-0002-0192-9351 (Jiaqing Yuan)
0000-0003-2971-5213 (Sangrong Sun)
0000-0001-5345-2594 (Hongling Ding)
0000-0002-1045-3818 (Xueqian Duan)
0000-0002-5769-7430 (Shaoqi Shen)
0000-0003-3430-9367 (Yanshuang Shen)
0000-0002-7772-7676 (Jing Li)
0000-0002-6042-090X (Fanbo Meng)
0000-0002-8166-7071 (Yangqin Xie)
0000-0002-0442-9205 (Jianyu Wang)
0000-0001-6786-2236 (Yue Hou)
0000-0002-3954-5038 (Jin Zhang)
0000-0003-3425-1011 (Xianchun Zhang)
0000-0001-7740-0932 (Xiu-Qing Li)
0000-0003-2159-0487 (Andrew H.Paterson)
0000-0003-3454-0374 (Xiyin Wang)
 Genomics,Proteomics & Bioinformatics2020年3期
Genomics,Proteomics & Bioinformatics2020年3期
- Genomics,Proteomics & Bioinformatics的其它文章
- CircPlant:An Integrated Tool for circRNA Detection and Functional Prediction in Plants
- Genome Assembly and Pathway Analysis of Edible Mushroom Agrocybe cylindracea
- Genome Size Evolution Mediated by Gypsy Retrotransposons in Brassicaceae
- Ubiquitinome Profiling Reveals the Landscape of Ubiquitination Regulation in Rice Young Panicles
- Characterization of Lysine Monomethylome and Methyltransferase in Model Cyanobacterium Synechocystis sp.PCC 6803
- Na2CO3-responsive Photosynthetic and ROS Scavenging Mechanisms in Chloroplasts of Alkaligrass Revealed by Phosphoproteomics