
Ubiquitinome Profiling Reveals the Landscape of Ubiquitination Regulation in Rice Young Panicles
2020-03-04 09:21:22LiyaZhuHanChengGuoqingPengShuansuoWangZhiguoZhangErdongNiXiangdongFuChuxiongZhuangZexianLiuHaiZhou
Liya Zhu,Han Cheng,Guoqing Peng,Shuansuo Wang,Zhiguo Zhang,Erdong Ni,Xiangdong Fu,Chuxiong Zhuang,Zexian Liu,Hai Zhou,
1 State Key Laboratory for Conservation and Utilization of Subtropical Agro-bioresources,Instrumental Analysis and Research Center,Key Laboratory of Plant Functional Genomics and Biotechnology of Guangdong Provincial Higher Education Institutions College of Life Sciences,South China Agricultural University,Guangzhou 510642,China
2 Sun Yat-sen University Cancer Center,State Key Laboratory of Oncology in South China,Collaborative Innovation Center for Cancer Medicine,Guangzhou 510060,China
3 School of Life Sciences,Zhengzhou University,Zhengzhou 450001,China
4 College of Life Science and Technology,Huazhong University of Science and Technology,Wuhan 430074,China
5 The State Key Laboratory of Plant Cell and Chromosome Engineering,Institute of Genetics and Developmental Biology,Chinese Academy of Sciences,National Centre for Plant Gene Research,Beijing 100101,China
Abstract Ubiquitination,an essential post-transcriptional modification (PTM),plays a vital role in nearly every biological process,including development and growth.Despite its functions in plant reproductive development,its targets in rice panicles remain unclear.In this study,we used proteome-wide profiling of lysine ubiquitination in rice(O.sativa ssp.indica)young panicles.We created the largest ubiquitinome dataset in rice to date,identifying 1638 lysine ubiquitination sites on 916 unique proteins.We detected three conserved ubiquitination motifs,noting that acidic glutamic acid (E) and aspartic acid (D) were most frequently present around ubiquitinated lysine.Enrichment analysis of Gene Ontology (GO) annotations and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of these ubiquitinated proteins revealed that ubiquitination plays an important role in fundamental cellular processes in rice young panicles.Interestingly,enrichment analysis of protein domains indicated that ubiquitination was enriched on a variety of receptorlike kinases and cytoplasmic tyrosine and serine-threonine kinases.Furthermore,we analyzed the crosstalk between ubiquitination,acetylation,and succinylation,and constructed a potential protein interaction network within our rice ubiquitinome. Moreover,we identified ubiquitinated proteins related to pollen and grain development,indicating that ubiquitination may play a critical role in the physiological functions in young panicles.Taken together,we reported the most comprehensive lysine ubiquitinome in rice so far,and used it to reveal the functional role of lysine ubiquitination in rice young panicles.
KEYWORDS Lysine ubiquitination;Ubiquitinome;Rice;Grain development;Male sterility
Introduction
Protein post-translational modifications (PTMs) regulate a abroad range of biological processes such as cell cycle,metabolism,and signal transduction[1-3].Among PTMs,ubiquitination,acetylation,methylation,and succinylation occur at lysine residue [4-7].PTMs play a vital role in regulating the activity,localization,and degradation of proteins.Ubiquitin is a small,highly conserved protein consisting of 76 amino acids.Ubiquitination depends on ubiquitin-activating enzyme(E1),ubiquitin-conjugating enzyme (E2),and ubiquitin ligase(E3) [8,9].Target proteins can be modified through mono-or multi-ubiquitination at different lysine sites by ubiquitin[10,11].Since the N-terminal methionine and seven lysine residues in ubiquitin could be ubiquitinated,this protein itself allows for the formation of various polyubiquitin chains [12].Different kinds of ubiquitination play important roles in diverse cellular processes,such as histone regulation,innate immune signaling,DNA repair,and endocytosis [9,13,14].
Progress in methods for the enrichment of ubiquitinated peptides and high-throughput mass spectrometry (HTP-MS)-based identification techniques greatly improves the identification of ubiquitination events [15,16].In 2003,Peng et al.[15]published a novel proteomics approach for identification of ubiquitin conjugates inSaccharomyces cerevisiaelysate,and detected 110 ubiquitination sites.In 2008,Meierhofer et al.[16]identified 669 ubiquitinated substrates and 44 ubiquitin attachment sites in HeLa cells.Although these studies have identified a number of ubiquitinated proteins,the number of mapped ubiquitination sites is limited due to the enrichment of ubiquitinated peptides.An enrichment method based on di-Gly-lysine-specific antibody allows to study ubiquitination signaling and to detect massive ubiquitination sites [17-21].For example,Wagner et al.[21]identified 11,054 ubiquitination sites on 4273 proteins in human cells by combining single-step immunoenrichment of modified peptides with peptide fractionation and HTP-MS.In 2017,global analysis of proteomes and ubiquitinomes revealed the involvement of ubiquitination in the degradation of proteins in petunia following ethylene treatment,identifying 2270 ubiquitinated sites in 1221 proteins in ethylene-mediated corolla senescence [20].
Rice(Oryza sativaL.)is a staple food source for about half of the global population and one of the most important crops in the world [22].Due to the extensive research of functional genomics,rice represent a model organism for monocot research on breeding and fundamental biology [23].With the completion of cultivated and wild rice genome sequences,as well as continuous refining of gene annotation,proteomic profiling stands as a powerful tool to investigate biological processes in rice.For example,isobaric tags for relative and absolute quantification (iTRAQ) comparison of heat-tolerant and-sensitive rice lines at early milky stage identified 38 differentially expressed proteins [24],while HTP-MS-based proteomic and phosphoproteomic analyses identified 4984 proteins and 3203 phosphorylated proteins with 8973 phosphorylation sites in developing rice anthers around the time of meiosis [25].Through combination of high-accuracy nano liquid chromatography-mass spectrometry/mass spectrometry(LC-MS/MS) with enrichment of acetylated and succinylated peptides,simultaneous analyses of lysine acetylation and succinylation during rice seed germination identified 699 acetylated sites in 389 target proteins,and 665 succinylated sites in 261 substrates[26].Moreover,global analysis of lysine ubiquitination in rice young leaves identified the first ubiquitinome in rice,consisting of 861 ubiquitinated peptides from 464 proteins,revealing a widespread regulatory role for this modification [19].However,compared to the number of identified ubiquitinated sites and target proteins in mammals,a larger scale of ubiquitinated sites and substrates in rice tissues are likely still to be detected.Therefore,it is necessary to conduct a proteome-scale analysis of ubiquitinated sites and substrates for exploring the modulation effect of ubiquitination in rice.
Hybrid rice has a 10%-20% yield advantage over the nonhybrid rice.Male sterility plays a critical role in hybrid breeding.Young panicles mark rice reproductive growth,and the shift from microsporocyte to meiosis stage is critical for rice reproductive development.Moreover,this event is particularly sensitive to the environmental stress [27-30].Previously,we found thatthermos-sensitive genic male sterile 5(tms5)-encoded RNase ZS1processes the mRNA of theubiquitin-60S ribosomal protein L40(UbL40) gene,controlling thermos-sensitive genic male sterility (TGMS) in rice [31].In this work,we performed a proteome-scale study of lysine ubiquitination in the young panicle tissue of rice(O.sativassp.indica),and identified a total of 1638 ubiquitinated sites in 916 unique proteins,representing the largest dataset of ubiquitinome in rice to date.Bioinformatical analysis indicated that ubiquitination in rice plays a critical role in different cellular processes.Taken together,our work not only identified the most comprehensive lysine ubiquitinome in rice so far,but also systematically indicated the function of lysine ubiquitination in rice young panicles.
Results and discussion
Large-scale profiling of ubiquitination sites in rice young panicles
For the purpose of acquiring the detailed landscape of lysine ubiquitination events in rice panicles,we extracted proteins from rice(O.sativassp.indica) young panicles at pollen mother cell and meiosis stages.After digestion with trypsin,the lysineubiquitinated peptides were enriched through affinity purification with ubiquitinated-lysine antibodies and analyzed by LC-MS/MS.Then,the LC-MS/MS-isolated lysine-ubiquitinated peptides were searched against the UniProt database.The mass errors for precursor ions and fragment ions were set to 6 ppm and 0.02 Dalton(Da),respectively.We identified a total of 1612 lysineubiquitinated peptides from 916 unique proteins with peptide score >40 (Table S1).Ubiquitination was not uniformly distributed in peptides of different lengths(Figure 1A).For example,the percentage of identified ubiquitinated peptides with lengths of 12,13,and 14 amino acids were 9.31% (150/1612),9.80%(158/1612),and 9.86% (159/1612),respectively.Furthermore,the majority of identified peptides had a single ubiquitination site(98.20%,1583/1612).However,there were also 29 peptides which included at least two ubiquitinated sites (Figure 1B).Approximately 64.08%of the identified proteins(587/916)contained a single ubiquitinated site,18.56% (170/916) carried two sites,and 8.73% (80/916) carried three (Figure 1C).Generally,the average degree of detected ubiquitination events was 1.79 (1638/916).We compared our data with another ubiquitinome profiled in young leaves ofO.sativassp.japonica[19].Using orthologous mapping,we found that nearly 33.76%(263/779)of ubiquitination sites and 47.80% (206/431) of ubiquitinated proteins detected in young leaves were also present in young panicles(Figure 1D).However,1375 ubiquitination sites and 710 ubiquitinated proteins were uniquely identified in our work.Taken together,our work identified the largest dataset of ubiquitinome in rice to date.

Figure 1 Characteristics of the identified lysine-ubiquitinated peptides in rice young panicles
The sequence and structure preferences of ubiquitination in rice young panicles
To evaluate the sequence preferences of the lysine ubiquitination in rice young panicles,we used Motif-x,developed to extract overrepresented patterns through comparison with a dynamic background [32],to search for the putative ubiquitination motifs in the identified ubiquitination dataset.We identified three conserved motifs significantly enriched in the ubiquitinome of rice young panicles,including E-Kub,Kub-D,and E-X-X-X-Kub,where Kubrepresents the ubiquitinated lysine residue andXrepresents any amino acid residue(Figure 2A).Motif analysis revealed that acidic glutamic acid(E) and aspartic acid (D) occurred most frequently around the ubiquitinated lysine residues in rice young panicles (Figure 2A and B).Moreover,E-Kubis also present in datasets from rice young leaves [19]and petunia corollas [20],and Kub-D and E-X-X-X-Kubexist in petunia corollas [20].This indicated that lysine ubiquitination is highly conserved in plants.
Furthermore,in order to check the relative abundances of different amino acids surrounding the ubiquitinated lysine sites,we built an heatmap for the 13-mers [33](Figure 2C),finding that alanine (A) was enriched around the modified lysine.Moreover,E was the residue with the highest frequency in position -1,which was also significantly enriched in positions-3 and-4.These results indicated that the heatmap data on sequence preferences matched our motif analysis.
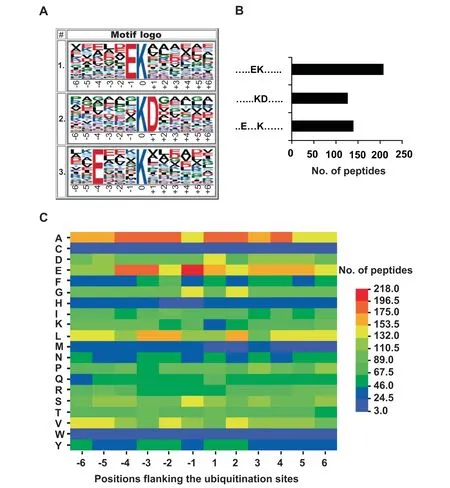
Figure 2 Motif analysis of ubiquitination sites in rice young panicles
To further dissect the recognition preferences of ubiquitination in rice,we performed a comparative analysis on sequential and structural features of the 1638 identified ubiquitinated lysine sites and the 24,346 non-ubiquitinated lysine sites.The position distribution of these datasets indicated that ubiquitination sites preferentially occurred in the central part of proteins,and the percentage of ubiquitination sites in Nterminus was higher than that of non-ubiquitination sites(Figure 3A).In addition,the distribution of secondary structures of the ubiquitination and non-ubiquitination sites demonstrated that both were enriched in coiled-coils,with an average coverage of approximately 53% (Figure 3B).Moreover,surface accessibility suggested that protein ubiquitination occurred preferentially on exposed lysine residues (Figure 3C)in protein ordered regions (Figure 3D).
Functional enrichment analysis of the identified ubiquitinated proteins
To better understand the biological functions of ubiquitination in rice young panicles,we annotated the identified ubiquitinated proteins through Gene Ontology(GO)enrichment analysis.GO annotations were significantly enriched in protein modification processes including protein phosphorylation (Pvalue=4.03E-09),protein deubiquitination (Pvalue=1.4 4E-04),and cellular protein modification process (Pvalue=1.44E-04) (Figure 4A and Table S2).Moreover,nucleosome assembly (Pvalue=8.18E-04),sucrose metabolic process(Pvalue=1.44E-03),chromosome organization (Pvalue=9.53E-03),and tricarboxylic acid cycle (Pvalue=9.53E-03) were also enriched in our ubiquitinome.Besides,ubiquitinated proteins were also found to be involved in multiple biosynthetic processes including cutin,wax,cytidine triphosphate (CTP),guanosine triphosphate (GTP),and uridine triphosphate (UTP) (Table S2).The most significantly enriched molecular functions were associated with adenosine triphosphate (ATP) binding (Pvalue=2.46E-13),protein serine/threonine kinase activity(Pvalue=2.16E-07),protein tyrosine kinase activity (Pvalue=1.72E-06),nucleotide binding (Pvalue=1.01E-04),and ubiquitin-protein transferase activity (Pvalue=1.44E-04) (Figure 4B and Table S2).The most significant cellular components were related to membrane,proteasome core complex,actin filament,and exine,indicating the most likely localization of the identified ubiquitinated proteins (Figure 4C and Table S2).On the basis of our enrichment analysis of GO terms,we supposed that the identified ubiquitinated proteins participate in a variety of biological processes and have diverse molecular functions in rice.
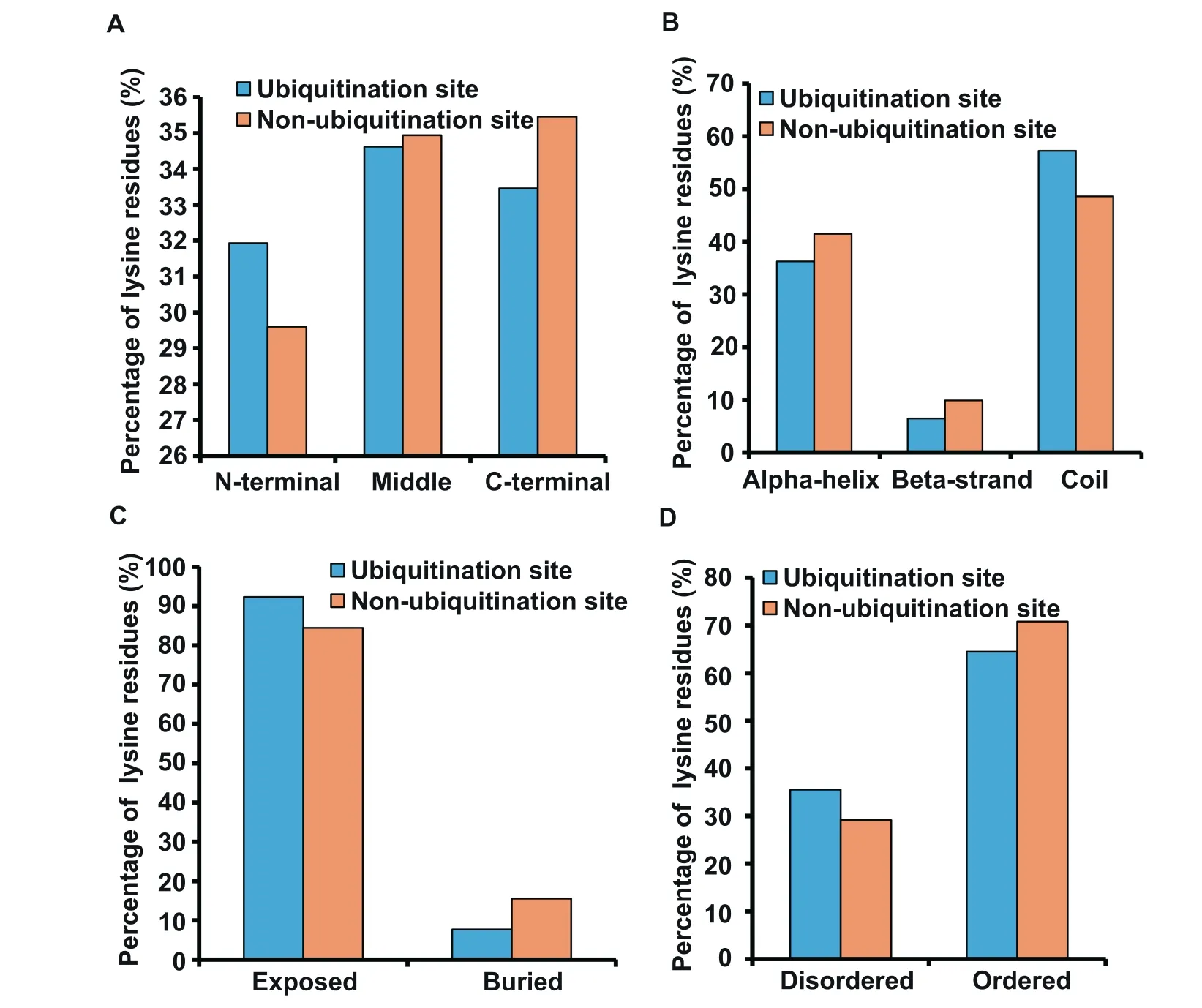
Figure 3 The preferences for sequence and structure of ubiquitination sites
To better understand which pathways ubiquitination regulated in rice,we carried out an enrichment analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in the ubiquitinome (Figure 5A and Table S3).The results showed that the KEGG pathways were most significantly enriched in proteasome (Pvalue=2.49E-06),ribosome (Pvalue=4.45E-05),phagosome (Pvalue=4.27E-04),protein processing in endoplasmic reticulum (Pvalue=1.25E-03),alongside with other biosynthesis and metabolism pathways.These results suggested that our ubiquitinome contains proteins involved in a large number of fundamental cellular processes critical for anther development in rice young panicles,such as protein degradation and protein quality control,DNA repair,innate immune signaling receptor,and endocytosis.
Since domains are the conserved parts of protein sequences and often form functional units,we used statistical analysis to identify enriched domains in the rice ubiquitinome.Among the identified ubiquitinated proteins,we observed that 150 protein domains were significantly enriched(P<0.01),including protein tyrosine kinase (Pkinase_Tyr),protein kinase domain(Pkinase),leucine rich repeat N-terminal domain(LRRNT_2),UBA protein domain (UBA),leucine rich repeats (2 copies)(LRR_4),leucine rich repeats (LRR_8),and ubiquitin (Figure 5B and Table S4).Protein ubiquitination plays a key role for the regulation of cell-surface receptors [34].In our rice ubiquitinome,many substrates were present on cell-surface receptors.Previous research has indicated that protein ubiquitination mediates the endocytosis of receptor tyrosine kinases(RTKs) and controls the extent and duration of receptor signaling in animals [35].Receptor-like kinases (RLKs) in plants are a class of transmembrane protein kinases whose basic structure is similar to that of RTKs [36].We indeed detected ubiquitinated RLKs containing domains including Pkinase_Tyr,LRRNT_2,LRR_4,or LRR_8 (Table S4).Moreover,ubiquitinated sites were identified on numerous cytoplasmic tyrosine and serine-threonine kinases which were involved in the transduction of signals from cell-surface receptors (Table S4).These data suggested that protein ubiquitination plays important roles in signal transduction of rice anther development.
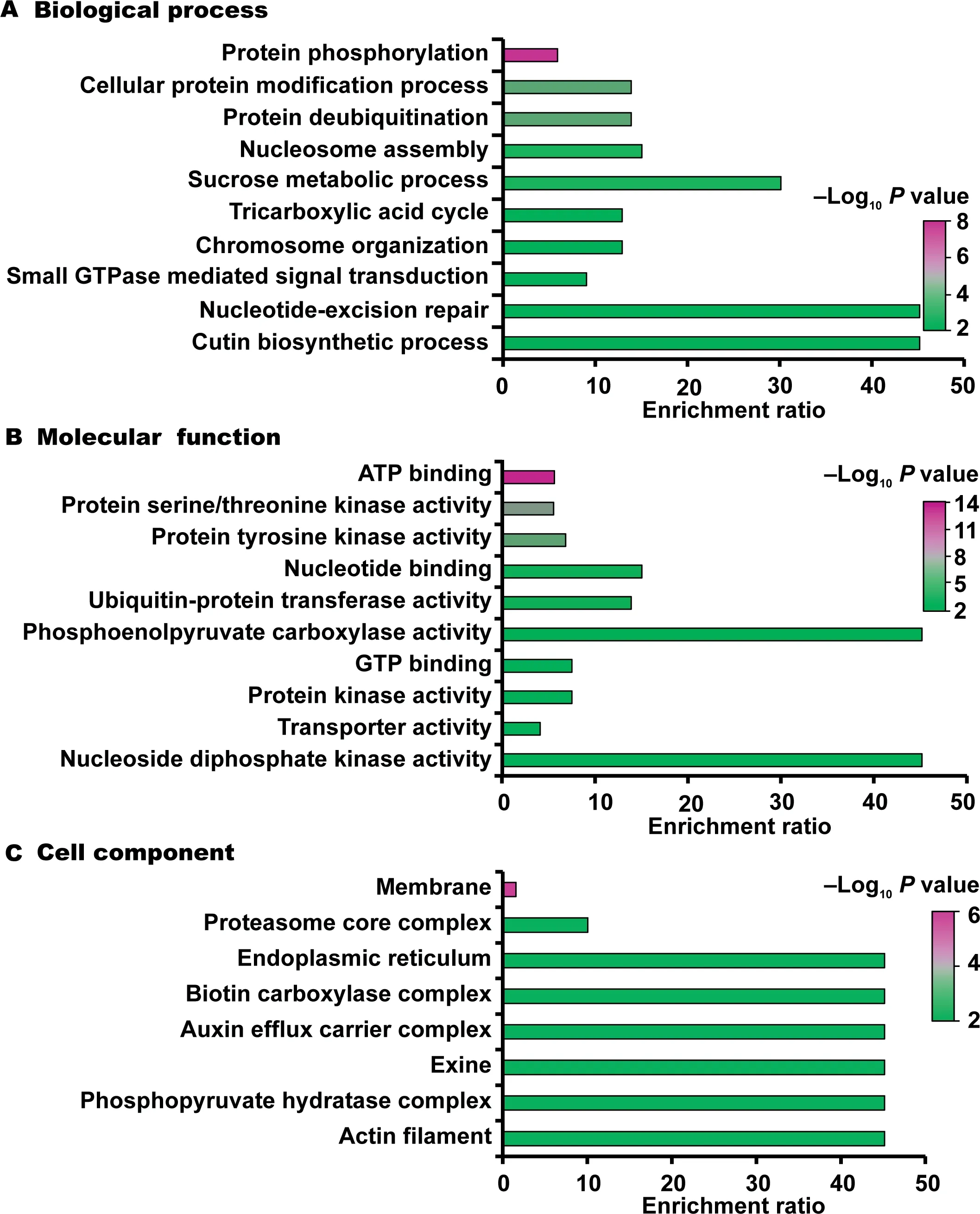
Figure 4 Enrichment analysis of GO annotations in identified ubiquitinated proteins (top 10)
Crosstalk between ubiquitination,acetylation,and succinylation
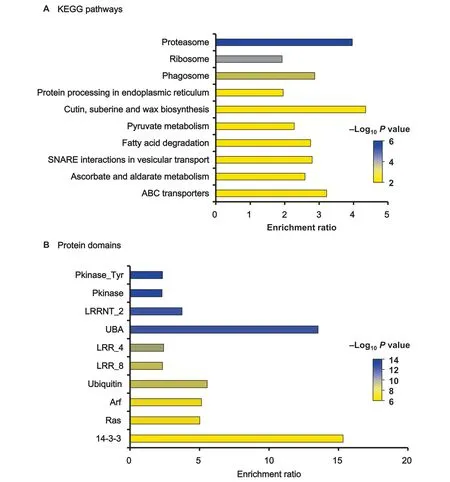
Figure 5 Enrichment analysis of KEGG pathways and protein domains in identified ubiquitinated proteins (top 10)
Besides ubiquitination,PTMs such as acetylation,succinylation,methylation,and SUMOylation are present on lysine residues [37],allowing PTMs to form complex regulatory mechanisms.For example,ubiquitination and acetylation can synergistically orchestrate protein activity and specific processes[38].Here,to analyze the overlap of ubiquitination with acetylation and succinylation in rice,we integrated experimentally identified acetylation and succinylation datasets injaponicafrom previously published literatures as listed on the literature database PubMed [26,39,40].Using orthologous mapping,we obtained 1316 acetylation sites in 693 target proteins,and 652 succinylation sites in 268 substrates.We compared ubiquitination sites identified here with the integrated acetylation and succinylation sites.We found that 5.6%(74/1316) of acetylated lysines and 19.8% (137/693) of acetylated substrates could be ubiquitinated (Figure 6A and B).The overlap between ubiquitination and acetylation sites in rice was lower than the 30% previously reported in human[21].This may be due to the lack of large-scale acetylation studies in rice (O.sativassp.indica),as we used acetylation data fromjaponica.To characterize proteins predicted to be both ubiquitinated and acetylated,we performed an enrichment analysis of KEGG pathways (Table S5).The pathways for ribosome(Pvalue=7.55E-08),proteasome(Pvalue=4.08E-04),carbon fixation in photosynthetic organisms (Pvalue=1.46E-03),glycolysis/gluconeogenesis (Pvalue=8.34E-03),and other metabolism pathways were significantly enriched among these proteins.In fact,a previously report also showed that Anaphase-promoting complex/cyclosome-cell division cycle 20-like protein 1 (APC/C-Cdh1) and SKP1/CUL-1/F-box protein-β-transducin repeats-containing proteins (SCF-β-TrCP),two ubiquitin ligases,sequentially regulate glycolysis during cell cycle in HeLa cells [41].These results indicated that ubiquitination plays an important role in the above pathways.Furthermore,we compared ubiquitination and acetylation sites during anther development in rice[42].Our data indicated that 9.5% (112/1181) of acetylated sites and 30.9% (188/609) of acetylated substrates could be ubiquitinated (Figure 6C and 6D).The overlap between ubiquitination and acetylation sites was higher than that in the integrated data (Figure 6A and B).The result suggested that the crosstalk between ubiquitination and acetylation in the same tissue and stage happens more frequently than that in others.Besides,the enrichment analysis of KEGG pathways indicated that this subset of proteins had a function in ribosome (Pvalue=1.79E-07),proteasome (Pvalue=5.11E-06),glycolysis/gluconeogenesis (Pvalue=1.26E-02),RNA transport (Pvalue=2.37E-02),carbon fixation in photosynthetic organisms (Pvalue=2.61E-02),and pyruvate metabolism (Pvalue=3.72E-02) (Table S6).
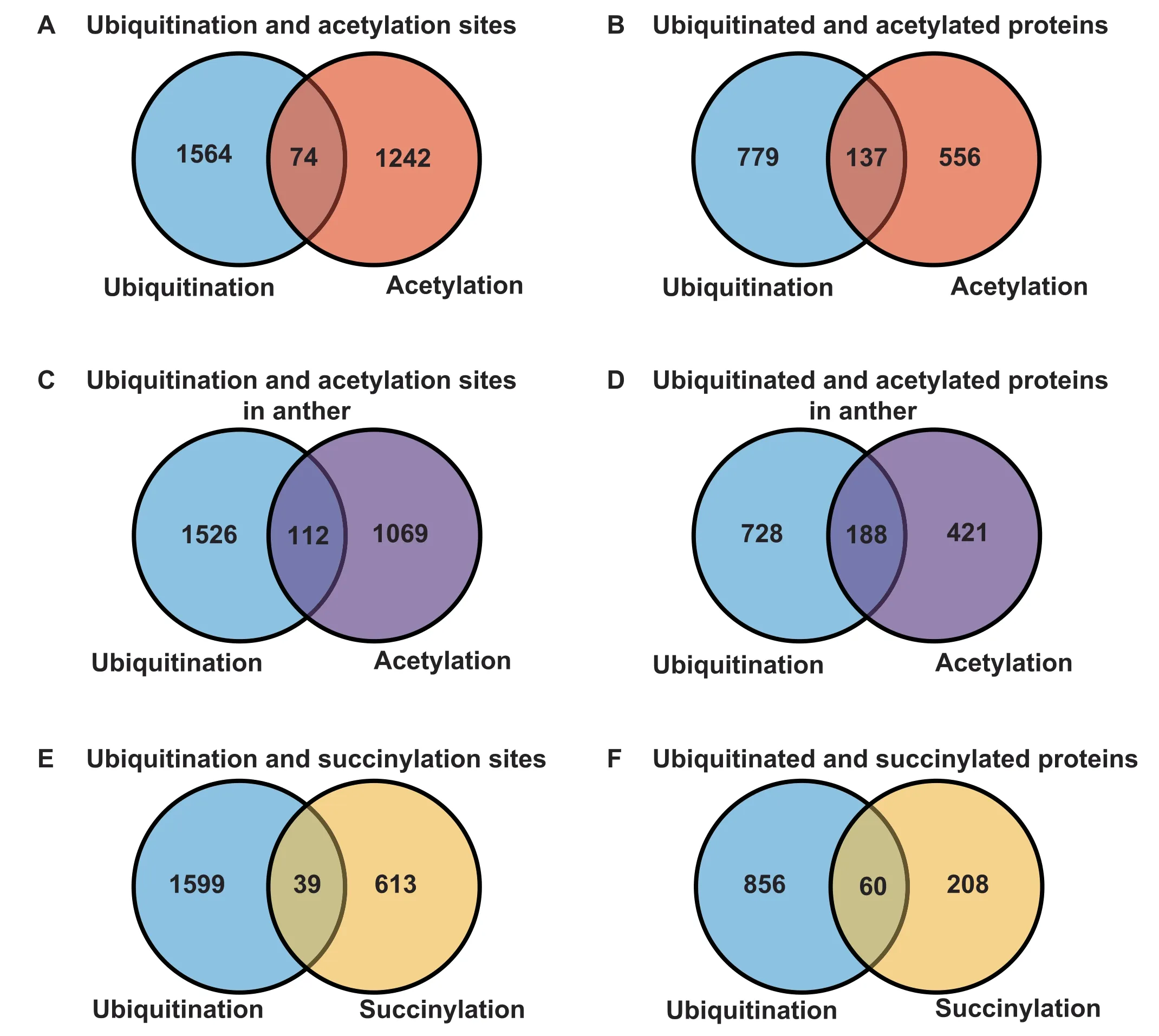
Figure 6 Summary of the overlap of ubiquitination in this study with acetylation and succinylation in the previously researches
Moreover,39(6.0%) succinylation sites were also ubiquitinated at the same position (Figure 6E),while 60 (22.4%) succinylated proteins were modified by ubiquitination(Figure 6F).Enrichment analysis of KEGG pathways suggested that the ubiquitination-succinylation crosstalk was significantly enriched in the pathways of glycolysis/gluconeogenesis (Pvalue=5.13E-05),carbon fixation in photosynthetic organisms (Pvalue=1.86E-04),ribosome (Pvalue=6.06 E-03),and other metabolism processes (Table S7),similar to the results for the crosstalk between ubiquitination and acetylation.
Protein interaction network analysis
The ubiquitination of proteins depends on E1,E2 and E3,which dynamically control protein function [43].Combining protein-protein interactions (PPIs) obtained from the search tool for the retrieval of interacting genes/proteins database(STRING,http://string-db.org) [44]and the information on E1,E2,and E3 extracted from the ubiquitin and ubiquitinlike conjugation database (UUCD,http://uucd.biocuckoo.org) [43],we constructed a potential network among E1,E2,E3 and substrates by Cytoscape[45],with 30,449 PPIs between 715 proteins,including 2 E1,8 E2,and 28 E3,and 677 substrates (Figure 7,Tables S8-S10).The interactions between E1/2/3 and ubiquitinated substrates are list in detail in Table S9.
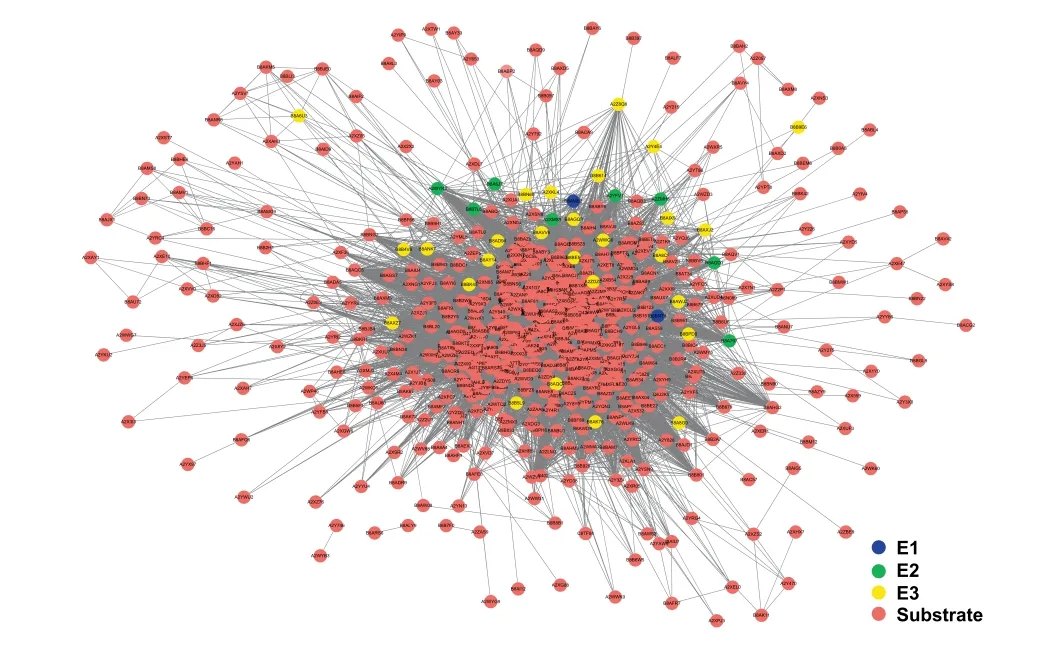
Figure 7 Interaction network of ubiquitinated proteins in rice young panicles
Moreover,we created protein clusters from the constructed PPI network via MCODE v1.4.2 in Cytoscape [46].The top 4 sub-networks with the highest scores are displayed inFigure 8.We also performed an enrichment analysis of KEGG pathways for the network proteins in each sub-network(Table S11).Statistical results demonstrated that the proteins in the networks were significantly associated with different pathways,including general metabolism,protein degradation,RNA biology,DNA damage repair,plant-pathogen interaction,autophagy,and innate immune signaling.This indicated that ubiquitinated proteins take part in a broad spectrum of PPIs in rice.
Functional analysis of lysine-ubiquitinated proteins involved in rice anther development
Ubiquitin homeostasis plays an important role in reproductive development [47].For example,the absence of the initiation codon ofpolyubiquitin 4(ubi4)results in the failure to undergo normal meiosis inSchizosaccharomyces pombe[48].Functional loss of thePolyubiquitin b(Ubb)in mice also leads to the arrest of spermatocytes and oocytes at the prophase of meiosis [49],and similar phenomena are also present inDrosophila[50].In order to investigate the role of ubiquitination during rice anther development,we compared protein-encoding genes detected in our lysine ubiquitinome and 410 genes preferentially expressed in early developing anther from previous transcriptomes [51].However,we failed to find any in our lysine ubiquitinome.This might be due to these anther-specific genes are essential for anther development at pollen mother cell and meiosis stages,or because their ubiquitination levels were too low for detection.We previously reported that excessive accumulation of ubiquitin resulted in rice male sterility at pollen mother cell and meiosis stages [31].
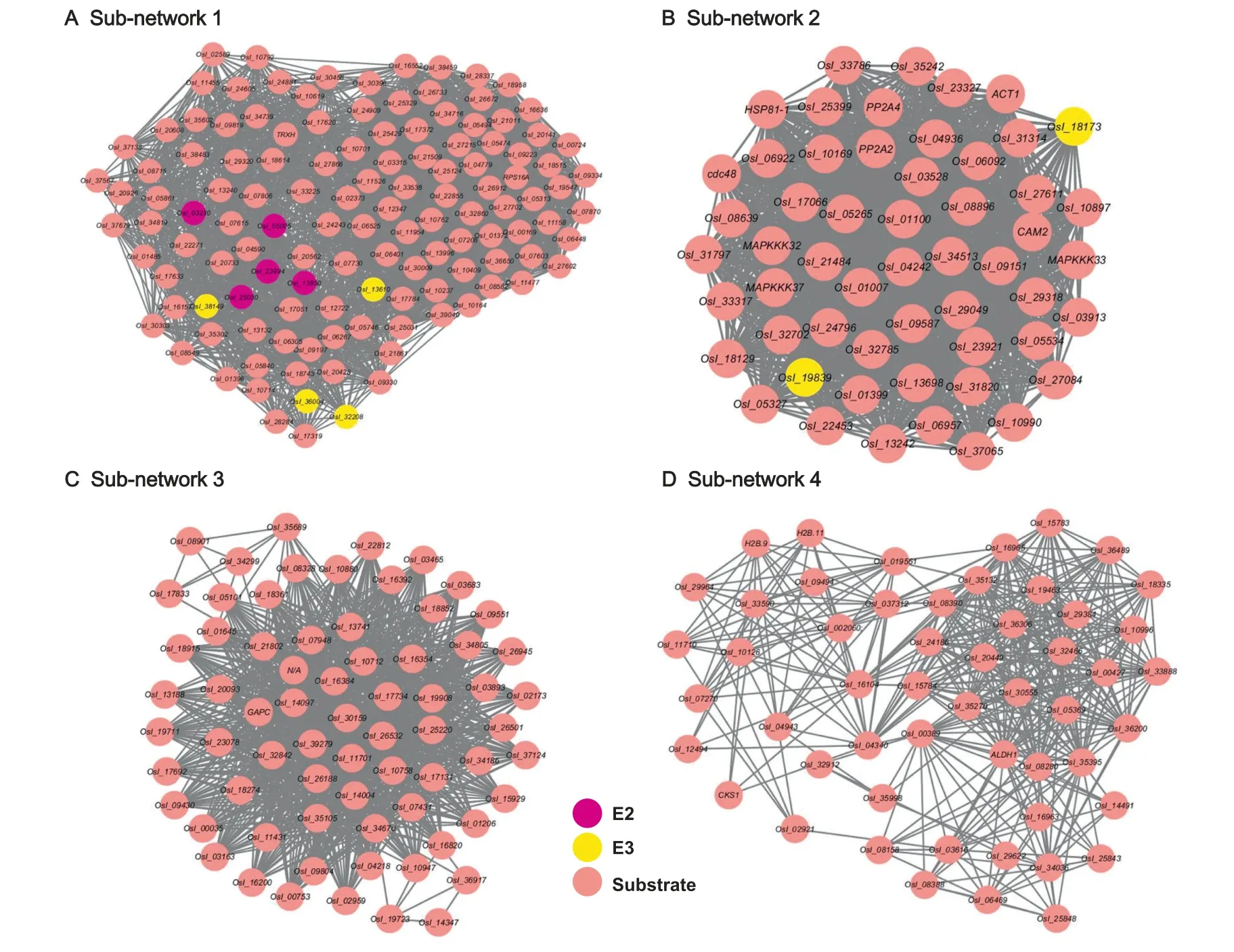
Figure 8 Top 4 sub-networks with the highest scores of rice young panicles as detected by MCODE v1.4.2

Next,we screened 106 anther development related proteins from the literatures(Table S12),and found that seven of them were present in our ubiquitinome dataset (Table 1).These seven proteins were Multiple Sporocyte 1 (MSP1) [52],UDParabinopyranose Mutase 3 (UAM3) [53],Wax-deficient Anther 1 (WDA1) [54],OsMADS7 [55],Narrow and Rolled Leaf 2 (NRL2) [56],Dwarf and Runtish Spikelet 1(OsDRUS1) [57],and OsDRUS2 [57].Except for OsDRUS1 and OsDRUS2,only one ubiquitination site was identified in each of the remaining five proteins.K11-and K48-linked polyubiquitination plays an important role in proteasome degradation [58,59],while K63-linked polyubiquitination may act on signal transduction,protein kinase activation,and endocytosis [60,61].Monoubiquitination is involved in a variety of cellular processes,such as histone regulation,gene expression regulation,DNA repair,and endocytosis [13,62-66].These findings suggested that the ubiquitination of anther development related proteins could be involved in anther development.Among these seven proteins,OsMADS7,OsDRUS1,and OsDRUS2 have functional redundancy,while the other four proteins are essential for anther development.Furthermore,MSP1 and WDA1 function in anther development during meiosis,while the other five proteins are responsible for early or late anther development.In addition,WDA1 and UAM3 only have effects on anther development,while the other five proteins affect not only anthers,but also the development of other organs in rice.These results suggested that most of the proteins essential for anther development might not be highly ubiquitinated when they are required at these stages,and that even the ubiquitinated proteins discovered in our study may not enter the ubiquitin proteasome system for degradation.In contrast,during the transition from the microsporocyte stage to the meiosis stage,various proteins may be degraded in time to ensure the succeed of anther development.As we know,the mutation of plant Ubox 4 (AtPUB4),a U-box/ARM repeat E3 ligase,leads to thermo-sensitive male sterility inArabidopsis[67].The substrate proteins for this E3 ligase might be among the identified ubiquitinome in this study.
Functional analysis of lysine-ubiquitinated proteins involved in rice grain development
The development of grain size has an important effect on rice yield.So far,of the 30 previously identified proteins related to the grain size in rice,three were in our ubiquitinome,including Small and Round Seed 5 (SRS5),Heading and Grain Weight(HGW),and New Plant Type 1(qNPT1)(Table 2).Five ubiquitination sites were identified in SRS5,while HGW and qNPT1 only had a single ubiquitination site.SRS5 is an alpha-tubulin protein [68],andsrs5mutants show small and round seed phenotypes.HGW is a plant-specific protein with an ubiquitin-associated (UBA) domain [69].It controls grain size and weight throughGrain Incomplete Filling 1(GIF1),Grain Size 3(GS3),Grain Weight 2(GW2),andGrain Width on Chromosome 5(GW5),and defectivehgwresults in small grain size.qNPT1 is a deubiquitinating enzyme with homology to human ovarian tumor domain-containing ubiquitin aldehyde-binding protein 1 (OsOTUB1),which inhibits the K63-linked ubiquitination of Ideal Plant Architecture 1(IPA1) but increases the K48-dependent degradation of IPA1[70].Furthermore,IPA1 Interacting Protein 1 (IPI1),a RING-finger E3 ligase,reduces protein abundance of IPA1 in panicles[71],and low levels of IPA1 reduce grain size in rice.These results indicated that ubiquitination plays an important role in the regulation of rice grain size,and can improve the rice yield by regulating the stability and activity of grain size related proteins.
Validating the ubiquitination of OsDRUS1 and OsOTUB1 in rice young panicles
To further validate the profiled ubiquitinome in rice young panicles,co-immunoprecipitation (Co-IP) and Western blot experiments were performed for proteins including OsDRUS1[57]and OsOTUB1[70],which were important for anther and grain development.As shown inFigure 9A,Myc-tagged OsDRUS1 (OsDRUS1-Myc) was detected in OsDRUS1-Myc expressed young panicles but not indrus1mutant.Furthermore,the pulled down OsDRUS1-Myc fusion proteins through anti-Myc antibody based Co-IP were detected as ubiquitinated by anti-ubiquitin antibody,and the low mobility bands could be corresponded to the addition of ubiquitin molecules.For OsOTUB1,the green fluorescent protein(GFP)-fused proteins were detected inOsOTUB1-GFPexpressed young rice panicles but not in the wild-type ones.Also,ubiquitination was detected by anti-ubiquitin antibodyfor the pulled down OsOTUB1-GFP proteins through anti-GFP antibody based Co-IP (Figure 9B).The high abundant band should be mono-ubiquitinated OsOTUB1-GFP,and the low abundant bands should be the polyubiquitinated OsOTUB1-GFP (Figure 9B).Taken together,these results confirmed the ubiquitination of OsDRUS1 and OsOTUB1 in rice young panicles and convinced the reliability of ubiquitinome profiling in this study.
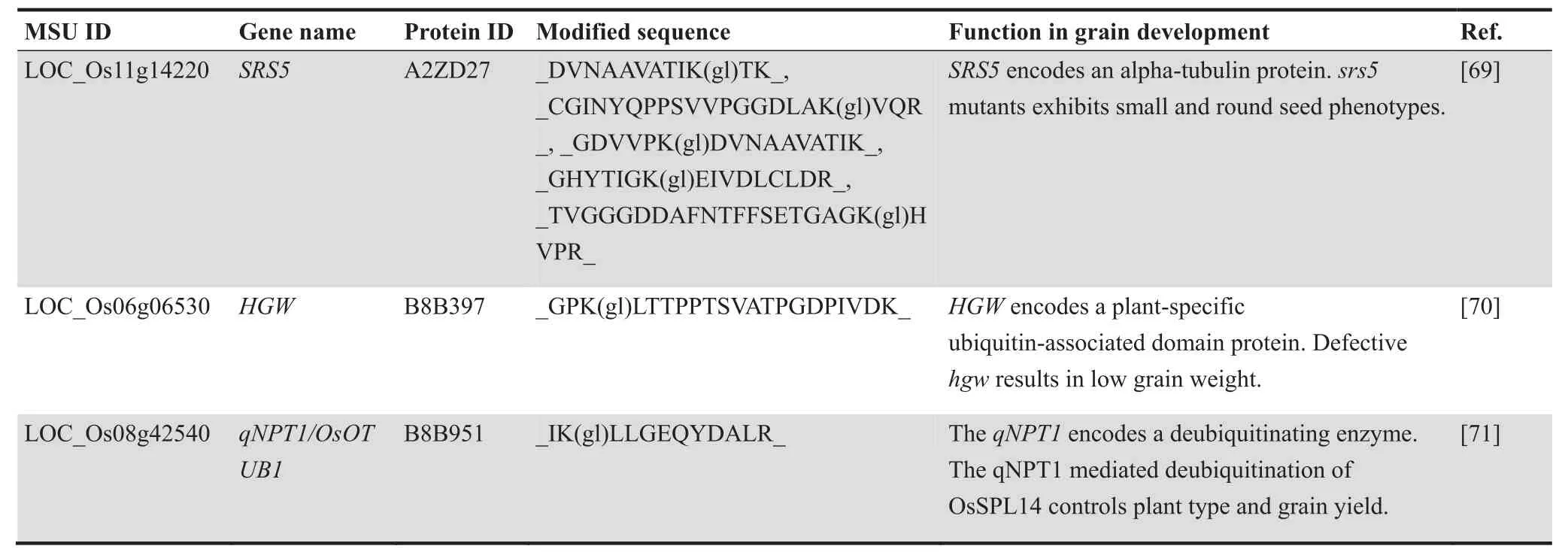
Table 2 Rice grain size related proteins identified in ubiquitinome
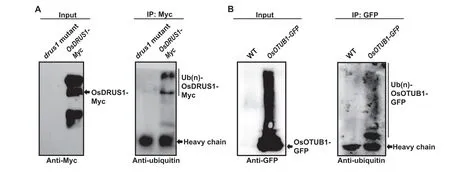
Figure 9 Validating the ubiquitination of OsDRUS1 and OsOTUB1 in rice young panicles
Conclusion
Ubiquitination plays a crucial role in nearly every biological process,and different ubiquitination sites may determine different fates of target proteins.In this study,we identified the largest ubiquitinome dataset in rice to date using on immune-affinity enrichment of ubiquitinated peptides and high-resolution mass spectrometry.Our ubiquitinome included 1638 lysine ubiquitination sites on 916 unique proteins.Functional characterization showed that ubiquitination plays a vital role in various biological processes,including anther and grain development.Furthermore,we constructed a potential protein interaction network among our ubiquitinome and analyzed the crosstalk between ubiquitination and acetylation or succinylation.We also detected three conserved ubiquitination motifs in rice panicles.Our ubiquitinome data not only enriched the catalog of ubiquitination sites in plants,but also that of candidate genes to study reproduction and grain development.These findings contribute to future studies on the regulation of ubiquitination in plants,especially in rice.
Materials and methods
Rice planting and isolation of samples
AnongN (O.sativassp.indica) is a sister line of thermossensitive genic male sterility line AnongS [31]and was planted in the growth chamber with 13.5-h light and 10.5-h dark under 28°C.Young panicles at pollen mother cell and meiosis stages were harvested for subsequent protein extraction.
Protein extraction and digestion
Young panicles were ground in liquid nitrogen,and the tissue powder was harvested into the centrifuge tube in lysis buffer(8 M urea,2 mM ethylene diamine tetraacetic acid (EDTA),10 mM dithiothreitol(DTT),50 μM PR-619,and 1%protease inhibitor cocktail).Sonication was performed three times on ice by a high-intensity ultrasonic processor (Scientz,Ningbo,China).Debris were removed through centrifugation at 20,000gat 4 °C for 10 min.The proteins were precipitated with 15%pre-cooled trichloroacetic acid (TCA) buffer under 4 °C for 2 h and washed with cold acetone three times while discarding the supernatant after 10 min centrifugation under 4 °C.Then the proteins were redissolved in buffer(100 mM tetraethylammonium bromide(TEAB),8 M urea,pH 8.0)and quantifiedwith the 2-D Quant kit(GE Healthcare,MA)according to the manufacturer’s instructions.The protein solution was reduced by 10 mM DTT for 1 h at 37 °C and alkylated through 20 mM indole-3-acetic acid(IAA)for 45 min under room temperature with darkness,followed by dilution through 100 mM TEAB of less than 2 M urea.Finally,trypsin was added according to the 1:50 trypsin/protein mass ratio for overnight digestion followed by a 1:100 trypsin/protein mass ratio for 4 h digestion.For each sample,approximately 100 mg proteins were digested with trypsin.
High performance liquid chromatography fractionation
The peptide samples were fractionated through high pH reverse-phase high performance liquid chromatography(HPLC) with Agilent 300 Extend C18 column (4.6 mm ID,5 μm particles,and 250 mm length).Peptides were first separated into 80 fractions through a gradient of 2% to 60% acetonitrile in 10 mM ammonium bicarbonate(pH 10)for 80 min.Then,the peptides were integrated into four fractions and dried by vacuum centrifuging.
Enrichment of lysine-ubiquitinated peptides
To enrich lysine-ubiquitinated peptides,peptides were dissolved in NETN buffer (100 mM NaCl,50 mM Tris-HCl,0.5% NP-40,1 mM EDTA,pH 8.0) and incubated with prewashed antibody beads (PTM Biolabs,Hangzhou,China)under 4°C with overnight gentle shaking.The beads were then washed four times in NETN buffer and twice in ddH2O.The peptides bound to beads were eluted in 0.1% trif luoroacetic acid (TFA),and the eluted fractions were vacuum-dried.The collected peptides were also cleaned by C18 ZipTips (Millipore,MA).
LC-MS/MS analysis
In previous studies [20],three parallel LC-MS/MS analyses were carried out for each fraction.A reverse-phase precolumn(Acclaim PepMap 100;ThermoFisher Scientific,MA) was used to load the dissolved peptides in 0.1% fatty acids,and a reverse-phase analytical column(Acclaim PepMap rapid separation liquid chromatography;ThermoFisher Scientific) was used to separate the peptides.The gradient of solvent B(0.1% fatty acids in 98% acetonitrile) included 8% to 25%for 26 min,25% to 38% for 8 min,38% to 85% for 4 min,and holding at 85% for 4 min.The flow rate was 280 nl/min on the ultra-performance liquid chromatography system(EASY-nLC 1000).The tandem mass spectrometry (MS/MS)of Q Exactive Plus Hybrid Quadrupole-Orbitrap (Thermo-Fisher Scientific) was coupled online to the ultraperformance liquid chromatograph after the peptides were subjected to the nanoSpray ionization source.The normalized collision energy was set as 30,and the resolution was set as 70,000 and 17,500 for the Orbitrap to detect the intact peptides and ion fragments,respectively.The data-dependent procedure was carried out in the mass spectrometry survey scan with 30 s dynamic exclusion as one MS scan followed by 20 MS/MS scans for the top 20 precursor ions above the threshold ion count of 1.5E-4.The electrospray voltage was set as 2 kV.Through automatic gain control,overfilling of the ion trap was prevented,and 1.5E-4 ions were accumulated to generate MS/MS spectra.For MS scans,the mass-to-charge ratio range was set as from 350 to 1800,and the fixed first mass for MS/MS was set as 100.
Database search
MaxQuant software suite with an integrated Andromeda search engine (version 1.3.0.5) [72]was employed to search against UniProt protein database (40,543 sequences) for identifying proteins and ubiquitination sites from tandem mass spectra,while sequences of reverse decoy and common contaminant proteins were used for false positive control.Trypsin/P was the cleavage enzyme,and up to 3 missing cleavages were allowed.Minimum peptide length was set at 7,and 4 modifications per peptide and 5 charges were allowed.Mass error was set as 6 ppm for precursor ions,and fragment ion tolerance was set as 0.02 Da.Carbamidomethylation on cystine was set as fixed modification,while oxidation on methionine,and ubiquitination on lysine and protein Nterminal methionine were set as variable modifications.The thresholds of false discovery rate (FDR) for the identification of proteins,peptides and modification sites were set at 1%.Default settings in MaxQuant were adopted for the other parameters.The identified potential lysine ubiquitination sites with the localization probability less than 75% or from the reverse or contaminant sequences were deleted.
Ubiquitination motif analysis
Motif-x[32]was employed to identify potential ubiquitination motifs present in our ubiquitinome data of rice young panicles.13-mer peptides with six upstream and downstream residues flanking the ubiquitination sites were obtained.If the location of ubiquitination site in the protein was at N-terminus or C-terminus,the ubiquitinated peptide was complemented to 13-mers with necessary numbers of ‘‘*”instead of any amino acid.The parameters of‘‘pre-aligned”,central K,width=13,occurrences=20,and significance=0.000001 were employed,while the protein sequences of rice (O.sativassp.indica) in UniProt were uploaded as the background.
Sequential and structural analyses
From our ubiquitinome data,we extracted 1638 ubiquitination sites and 24,346 non-ubiquitinated lysine residues.To study the positional distribution of these sites in proteins,the sequences were classified into three sections in average including N-terminus,middle,and C-terminus.Then,NetSurfP version 1.1 was used to predict the secondary structure and surface accessibility for these ubiquitination sites and nonubiquitinated lysine residues [73].In addition,we employed the ESpritz to predict the disordered regions of the ubiquitinated proteins from their amino acid sequences for studying the region preference of protein ubiquitination [74].
The statistical enrichment analyses
The gene association files of GO (10/28/2016) were obtained from the Gene Ontology Consortium database(http://geneontology.org/) [75].Because the number of GO terms in rice (O.sativassp.indica)is still quite limited,we further integrated the GO annotations ofOryza.sativassp.japonicausing blastall[76].Mapping ubiquitinated proteins to the GO annotations resulted in 13,640 proteins in rice (O.sativassp.indica) annotated by at least one GO term,with 302 annotated ubiquitinated proteins.KEGG annotation was from purchased FTP(File Transfer Protocol)subscription for private use[77].However,there was no data of KEGG pathways in rice (O.sativassp.indica),thus by mapping to the KEGG pathways ofjaponicathrough BLAST,we obtained 4115 rice (O.sativassp.indica) proteins annotated by at least one KEGG term,which contained 278 ubiquitinated proteins.We also obtained 793 ubiquitinated proteins against 22,289 rice(O.sativassp.indica)proteins annotated by at least one entry of domains.Afterwards,the overrepresented functional distributions,enrichment pathways and domains of ubiquitinated proteins were statistically analyzed for hypergeometric distribution [78].
Co-IP and Western blot of ubiquitinated proteins
As previously described [31,70],proteins were extracted from young panicles (5-7 cm in length) using extraction buffer(0.05 M Tris-HCl,150 mM NaCl,5 mM EDTA,0.1% Triton X-100,0.2% NP-40,50 μM MG132,pH 7.5).Anti-GFP(Roche,Basel,Switzerland)or anti-Myc(GNI,Tokyo,Japan)antibody was added into the protein extraction (1-5 μg antibody/1 mg protein),which was incubated at 4 °C for at least 4 h.Agarose beads (Sigma-Aldrich,MO) were added (50 μl/ml) and incubated at 4 °C for at least 2 h,then washed using extraction buffer for 5 times and eluted with loading buffer.The eluted samples were isolated using sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membrane(Pall Corporation,NY).The membrane was enclosed by 5%skimmed milk powder,and was incubated at 37 °C for 2 h with anti-GFP,anti-Myc,or anti-ubiquitin(Millipore)antibody,followed by washing using TBST buffer for 5 times.The goat anti-rabbit IgG horseradish peroxidase (HPR) was used as the second antibody.The proteins were detected using electrochemiluminescence (ECL) solution and X-ray.
Data availability
The raw data and search files were made available at the iProX database (iProX:IPX0001099000).
CRediT author statement
Liya Zhu:Investigation,Resources,Validation,Writing-original draft.Han Cheng:Formal analysis,Writing -original draft,Funding acquisition.Guoqing Peng:Validation,Resources.Shuansuo Wang:Validation,Resources.Zhiguo Zhang:Resources.Erdong Ni:Resources.Xiangdong Fu:Supervision.Chuxiong Zhuang:Supervision.Zexian Liu:Conceptualization,Writing -original draft,Supervision,Funding acquisition.Hai Zhou:Conceptualization,Writing -original draft,Supervision,Funding acquisition.All authors read and approved the final manuscript.
Competing interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Acknowledgments
This manuscript has been edited by TopEdit.We thank Prof.Ying Sun (Hebei Normal University,China) for providing functional complement line(proDRUS1:DRUS1-7Myc-6His)ofOsDRUS1.This work was supported by the grants from the National Key Research and Development Program of China (Grant No.2016YFD0100400 to HZ),the National Natural Science Foundation of China (Grant Nos.31501069 to ZL,31601067 to HC,and 31571255 to HZ),the Guangdong Natural Science Funds for Distinguished Young Scholars,China (Grant No.2017A030306001 to HZ),the Pearl River S&T Nova Program of Guangzhou,China (Grant No.201710010106 to HZ),and the Scientific Research Project of Guangzhou,China (Grant No.201504010004 to CZ).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.01.005.
ORCID
0000-0002-6692-5466 (Liya Zhu)
0000-0002-6684-317X (Han Cheng)
0000-0001-6087-2861 (Guoqing Peng)
0000-0001-5922-4824 (Shuansuo Wang)
0000-0001-7825-4877 (Zhiguo Zhang)
0000-0001-5246-1763 (Erdong Ni)
0000-0001-9285-7543 (Xiangdong Fu)
0000-0001-7801-3432 (Chuxiong Zhuang)
0000-0001-9698-0610 (Zexian Liu)
0000-0003-3035-7038 (Hai Zhou)
 Genomics,Proteomics & Bioinformatics2020年3期
Genomics,Proteomics & Bioinformatics2020年3期
- Genomics,Proteomics & Bioinformatics的其它文章
- CircPlant:An Integrated Tool for circRNA Detection and Functional Prediction in Plants
- Genome Assembly and Pathway Analysis of Edible Mushroom Agrocybe cylindracea
- Paleo-polyploidization in Lycophytes
- Genome Size Evolution Mediated by Gypsy Retrotransposons in Brassicaceae
- Characterization of Lysine Monomethylome and Methyltransferase in Model Cyanobacterium Synechocystis sp.PCC 6803
- Na2CO3-responsive Photosynthetic and ROS Scavenging Mechanisms in Chloroplasts of Alkaligrass Revealed by Phosphoproteomics