
Genome Size Evolution Mediated by Gypsy Retrotransposons in Brassicaceae
2020-03-04 09:21:24ShiJianZhangLeiLiuRuolinYangXiangfengWang
Shi-Jian Zhang,Lei Liu,Ruolin Yang,Xiangfeng Wang,
1 Department of Crop Genomics and Bioinformatics,College of Agronomy and Biotechnology,National Maize Improvement Center of China,China Agricultural University,Beijing 100094,China
2 Beijing Key Laboratory of Plant Resources Research and Development,School of Sciences,Beijing Technology and Business University,Beijing 100048,China
3 College of Life Sciences,Northwest A&F University,Yangling 712100,China
Abstract The dynamic activity of transposable elements (TEs) contributes to the vast diversity of genome size and architecture among plants.Here,we examined the genomic distribution and transposition activity of long terminal repeat retrotransposons(LTR-RTs) in Arabidopsis thaliana(Ath)and three of its relatives, Arabidopsis lyrata (Aly), Eutrema salsugineum (Esa),and Schrenkiella parvula (Spa),in Brassicaceae.Our analyses revealed the distinct evolutionary dynamics of Gypsy retrotransposons,which reflects the different patterns of genome size changes of the four species over the past million years.The rate of Gypsy transposition in Aly is approximately five times more rapid than that of Ath and Esa,suggesting an expanding Aly genome.Gypsy insertions in Esa are strictly confined to pericentromeric heterochromatin and associated with dramatic centromere expansion.In contrast, Gypsy insertions in Spa have been largely suppressed over the last million years,likely as a result of a combination of an inherent molecular mechanism of preferential DNA removal and purifying selection at Gypsy elements.Additionally,species-specific clades of Gypsy elements shaped the distinct genome architectures of Aly and Esa.
KEYWORDS Brassicaceae;Comparative genomics;Gypsy retrotransposon;Copia retrotransposon;Genome size evolution
Introduction
Transposable elements (TEs) play important roles in shaping genome structure,directly or indirectly influencing gene function and creating novel genetic materials in host genomes[1,2].In plants,proliferation of long terminal repeat retrotransposons (LTR-RTs) through a ‘‘copy-and-paste”mechanism contributes to the vast diversity of genome size in different plant species [3].LTR-RTs are classified into two superfamilies based on the relative location of the integrase(INT) encoded by thepolgene,namely theGypsy-like andCopia-like retrotransposable elements (abbreviated as theGypsyandCopiaelements,respectively,hereafter),which have had different impacts on the evolution of genome size [4,5].Comparison of the recent activity of LTR-RTs,including their transposition rate and genomic context,among related plant species may facilitate estimation of trends in genome size changes over short periods of evolutionary time.Previous studies of the activity of LTR-RTs inArabidopsis lyrata(Aly) andArabidopsis thaliana(Ath) revealed active transposition of LTR-RTs inAlyand efficient removal of LTR-RTs inAth,suggesting that theAlygenome has likely expanded over the past five million years,while theAthgenome has likely shrunk during this time period [6].
TE activity may also influence gene family evolution.Retrotransposons mediate gene family expansion by carrying adjacent genes and incorporating them into other genomic locations during transposition.It has been reported that the genes of rice and sorghum captured by LTR-RTs and fixed in the population exhibit signatures of positive selection [7].Additional studies revealed the effects of natural selection on LTR-RTs.For example,Baucom and colleagues [8]systematically analyzed rice LTR-RTs and detected strong purifying selection,as well as a few episodes of positive selection.However,the scenarios in which positive selection may occur are poorly understood.
The patterns of LTR-RT integration into the host genome vary greatly among LTR-RT clades and involve specific LTRRT domains,as well as interactions between LTR-RTs and the host genome [9-11].The observed distribution pattern of LTR-RTs in a current genome might be a result of the combined effects of purifying selection and preferential insertion of LTR-RTs [12].A type of LTR-RTs that includes chromoviruses (specific lineages ofGypsyelements) and those within theAthilafamily is especially intriguing to researchers because it exhibits strong site-targeting specificity [13,14].Recently,Weber et al.[10]characterised the chromoviruses ofBeta vulgarisbearing specific types of chromodomains,facilitating preferential insertion of this type ofGypsyelements into designated locations in a host genome.
The Brassicaceae is an economically important family of angiosperms.With the exception of the invaluable model plantAth,many species in this family,includingEutrema salsugineum(Esa),Schrenkiella parvula(Spa),Aly,Brassica rapa,andCapsella rubella,have recently been sequenced or are scheduled to be sequenced [6,15-20],providing rich genomic resources suitable for comparative genomics studies.Most importantly,these species are native to highly diversified niches.For example,EsaandSpaare naturally salt-tolerant species that inhabit harsh environments,including soils of high salt concentration.A majority of LTR-RTs are recognised as transcriptionally quiescent during normal plant development,but are capable of strong activation by a variety of abiotic and biotic stresses [21,22].Excessive transposition of LTRRTs may damage genome integrity and lead to impaired gene function.To better understand the dynamics of LTR-RTs,host genomes and their co-evolution,we comprehensively identified and characterized the LTR-RTs ofEsaandSpaalong with two related salt-sensitive species,AthandAly.To identify the possible mechanisms underlying TE proliferation defence,we systematically compared the genomic composition,transposition activity,insertion patterns,genomic distribution and selective forces of LTR-RTs among the four genomes.
Results
Distinct activity of Gypsy elements in the four Brassicaceae species
The four wild species in the Brassicaceae family used for comparative analysis are of high relatedness,with over 90%of the protein-coding genes positioned in high genomic synteny,as illustrated by the comparison of different genomic elements betweenAlyandEsa(Figure 1).Because of their similar habitats and life cycles,these species are considered to be good model systems for investigation of miscellaneous non-genic elements in Brassicaceae.The genome sizes of the four Brassicaceae species in this study vary greatly:~250 Mb forEsa(8 chromosomes),~200 Mb forAly(7 chromosomes),~140 Mb forSpa(8 chromosomes),and~125 Mb forAth(5 chromosomes).
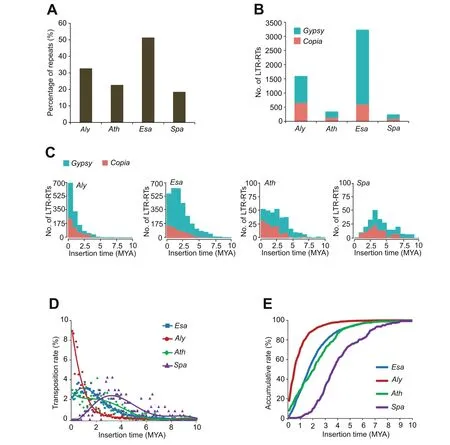
Figure 2 Different activity of LTR-RTs in the four genomes
As genome size diversity in plants is primarily influenced by TEs,we compared the total content of repetitive sequences in the four species[23].We found thatEsaandSpacontained the highest and lowest proportion of repeats (51.4% and 18.5%),respectively (Figure 2A).Accordingly,full-length LTR-RTs,which have retained the paired LTRs at the two ends and may still possess transposition ability,are most abundant inEsaand least abundant inSpa(Figure 2B).By estimating TE insertion times,we found that the burst(estimated by median age) of LTR-RTs inEsa(~2.5 million years ago;MYA)occurred later than inSpa(~4.1 MYA),but earlier than in bothAly(~0.7 MYA)andAth(~1.9 MYA)(Figure 2C).Additionally,the larger genomes ofEsaandAlyare mainly attributable toGypsyelements that might still actively transpose and are present at proportions significantly higher than those ofCopiaelements (Figure 2B).Whereas most insertions of LTR-RTs inEsa,Ath,andAlyare less than two million years(MYs)old,very few new insertions were found inSpa,suggesting very distinct evolutionary dynamics and mobile activity of these retrotransposable elements of the four species over the last few million years.
Genome size evolution,either expansion or shrinkage,is a dynamic process of DNA removalversusTE proliferation,with influence from,but not limited to,natural selection and inherent TE activity [23-25].Although the exact direction of species evolution over a long evolutionary history is difficult to determine,the evolutionary tendency can be inferred from the most recent transposition events of certain TE classes.To compare TE activity,we calculated the transposition rate as the net increase in the number of LTR-RTs within every 0.1 MYs over a 10-MY period (Figure 2D).Within the last three MYs,the transposition rate ofEsahas been relatively stable,whereas that ofSpahas been in continuous decline.In comparison,the transposition rate ofAlyhas increased continuously,whereas that ofAthhas remained relatively stable.The same tendency was also observed in terms of the accumulative rate of LTR-RTs within every 0.1 MYs over a 10-MY scale in the four genomes (Figure 2E).
Genomic distribution of the Gypsy elements in the four Brassicaceae species
We next compared the genomic distribution of full-length LTR-RTs among the four species and found that,whereasGypsyelements inAthandSpaare frequently present in centromeres,they are strongly localized to gene-poor pericentromeric heterochromatin inEsa(Figure 3A and Figure S1).As the length of euchromatic portions are similar inEsaandAth,the larger size of theEsagenome,half of which is comprised of pericentromeric heterochromatin,can be attributed to centromere expansion mediated byGypsyproliferation inEsa.By comparison,Gypsyinsertions inAlyappear more frequently in euchromatic regions without forming expanded pericentromeric heterochromatin (Figure S1).Additionally,the median distance betweenGypsyinsertions and their nearest genes is longer inEsathan inAly,whereas no such differences forCopiainsertions are apparent among the four species(Figure 3B).Strikingly,the medianGypsy-to-gene distance inSpais 0 kb,indicating that mostGypsyelements overlap with genes.This finding seemingly contradicts the assumption that TE insertions in genes are more deleterious than intergenic insertions.To investigate this pattern,we added insertion times to theGypsy/Copia-to-gene distance distribution in each species.Interestingly,each distribution exhibits primary and secondary peaks for bothGypsyandCopiaelements (Figure 3C).InEsa,Aly,andAth,the primary peaks represent intergenic insertions,and the secondary peaks,centred at 0 kb,are genic insertions that are predominantly younger(<1 MYA) than the intergenic insertions.InSpa,whereas 82%ofGypsyelements overlap with genes forming the primary peak,78% ofCopia-like elements are located in the intergenic regions forming the secondary peak.The contrast between the depletion of intergenicGypsyelements and the retention ofCopiaelements indicates that a strongly biased elimination ofGypsy-related sequences occurs inSpathrough unknown mechanisms,leading to the overall suppression ofGypsyactivity.This pattern supports our finding thatSpahas likely been downsizing its genome over the last two million years.
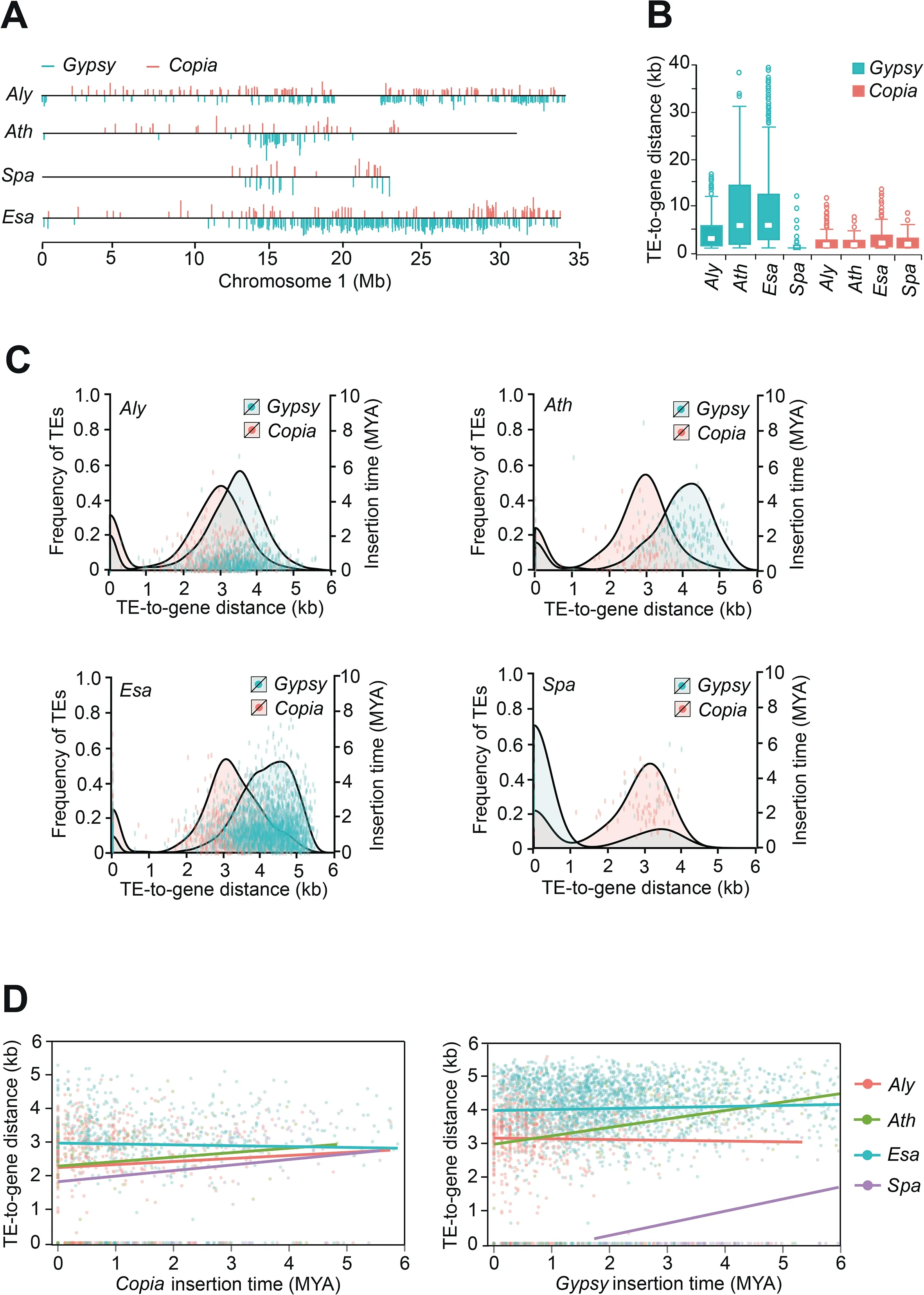
Figure 3 Genomic distribution of LTR-RTs in the four genomes
To determine whether the longerGypsy-to-gene distance inEsais a result of natural selection or inherent transposition bias,we analyzed the correlation between insertion times and insertion distances (Figure 3D).The magnitudes of the slopes of the regression lines are 0.028,0.033,0.056 and 0.105 forAly,Esa,Ath,andSpa,respectively.Whereas the most recent(<1 MYs)Gypsyinsertions inAthare predominantly located near genes,at distances similar to or slightly shorter than those observed inAly,there is an enrichment of olderGypsyinsertions at distances far from genes inAth.Interestingly,inEsa,we detected a statistically significant,weak correlation between theGypsy-to-gene distance and insertion time(P=6.75×10-3),and insertions inEsaare consistently farther away from genes than they are inAly.
Preferential DNA removal of Gypsy-related sequences in Spa
The absence of intergenicGypsyelements and lack of new insertions(<2 MYs)inSpasuggest an active role for the host genome in aggressively prohibitingGypsyproliferation,thereby leading to disproportionate predominance ofCopiaandGypsyelements.However,further investigation is required to determine whether the observed depletion ofGypsyelements inSpais caused by preferential DNA removal ofGypsyelements via an inherent molecular mechanism or purifying selection ofGypsyelements.DNA removal has been hypothesized to play a major role in preventing TE proliferation-mediated genome expansion [5,24,25].Full-length LTR-RTs with a pair of identical direct repeats (paired-LTRs) are particularly favoured for DNA removal via unequal homologous recombination (HR) events,because the two LTRs provide homologous sequences to initiate illegitimate recombination [25-27].Frequent HR-mediated DNA removal may result in a high abundance of solo-LTR remnants in the genome,which can be used as evidence to prove the existence of an inherently efficient DNA removal mechanism.
Thus,we compared the ratios of solo-LTRsversuspaired-LTRs ofCopiaandGypsyelements among the four genomes.The relative abundances of solo-and paired-LTRs were used to evaluate the propensity of HR-mediated removal of active LTR insertions in the four genomes.We found that the numbers of solo-LTRs ofCopiaelements were 2.98,1.53,6.92,and 1.60 times those of paired-LTRs inAly,Ath,Esa,andSpa,respectively;whereas the corresponding ratios for theGypsyelements were 5.09,8.22,8.03,and 17.76 times greater(Table 1).Because bothGypsyandCopiaelements possess identical direct repeats,the intra-chromosomal recombination mechanism may not distinguish the two superfamilies of the LTRs.Thus,the disproportion of the solo-and paired-LTRs betweenGypsyandCopiaindicates thatGypsyelements were removed more efficiently thanCopiaelements in all four species,suggesting that the effect ofGypsyelements on individual survival is more deleterious than that ofCopiaelements.In addition,Spahas a much greater proportion of solo-LTRs ofGypsyin comparison with those of the other three species,while the ratio of solo-LTRsversuspaired-LTRs inCopiaelements was comparable betweenSpaandAth.In summary,our results suggest thatSpamight possess a highly efficient,inherent molecular mechanism to purgeGypsyelements,probably through HR-mediated DNA removal,to accelerate the processes of depressing deleterious,activeGypsyinsertions.
Phylogenetic analysis showed diverse LTR-RT clades in the four species
BothGypsyandCopiaelements are composed of many clades that can be phylogenetically classified based on their reverse transcriptase (RT) sequences [8].We first categorized all of the full-length LTR-RTs into three groups,namely ‘‘a(chǎn)ctive”,‘‘inactive”and ‘‘fossil”,based on the coding integrity of their RT domains.Active LTR-RTs most likely possess the capacity to transpose,while inactive LTR-RTs may have lost this capacity because of premature stop codons or frame-shift mutations in the RT domains;the remaining LTR-RTs are considered to be fossil LTR-RTs because their RT domains are severely fragmented.In all four species,inactive and fossil LTR-RTs are present in high proportions.Alyhas the highest proportion(58%)of active LTR-RTs,whileSpahas the lowest(13%);EsaandAthhave proportions of active LTR-RTs between those of the other two species (Figure 4A and B).
We further classified the group of active LTR-RTs into clades based on pairwise similarities in their RT domains using an affinity propagation clustering algorithm.It’s worth noting that clade classification based on featured domain similarity is relatively a simplified means of classification,because different classes of LTR-RTs are usually embedded with each other,leading to multiple returns from one query and making family/subfamily assignment difficult.As a result,20Copiaclades(c1-c20)and 7Gypsyclades(g1-g7)were generated among all of the LTR-RT sequences in the four species.These clades were subsequently superimposed,with high consistence onto a phylogenetic tree generated by the neighbor-joining (NJ)method,indicating that the LTR-RT classification is reliable(Figure 4C and D).These clades varied greatly in size:the numbers of elements range from 7 to 79 for theCopiaclades and from 45 to 280 for theGypsyclades.The LTR-RTs of the four species exhibited different compositions among the clades (Figure 5A).For example,Gypsyclade g3 andCopiaclades c2,c5,c8,and c9 mainly occur inAly,which is the youngest of the four species,whereasCopiaclades c11,c18,and c19,as well asGypsyclades g1,g6,and g7,dominantly occur inEsa,indicating species-specific proliferation of LTRRT clades inAlyandEsa.In contrast,most of theGypsyelements inAthandSpabelong to the clade g5 and clade g2,respectively,indicating that the mobile activity of most LTRRTs is suppressed in these two species(Figure 5A).The diverse compositions of LTR-RT clades in the four genomes were unexpected.
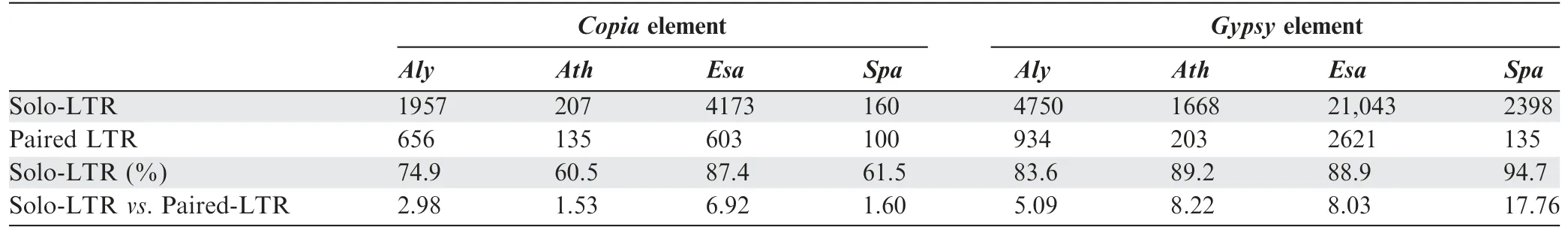
Table 1 Comparison of paired-and solo-LTRs of Copia and Gypsy elements
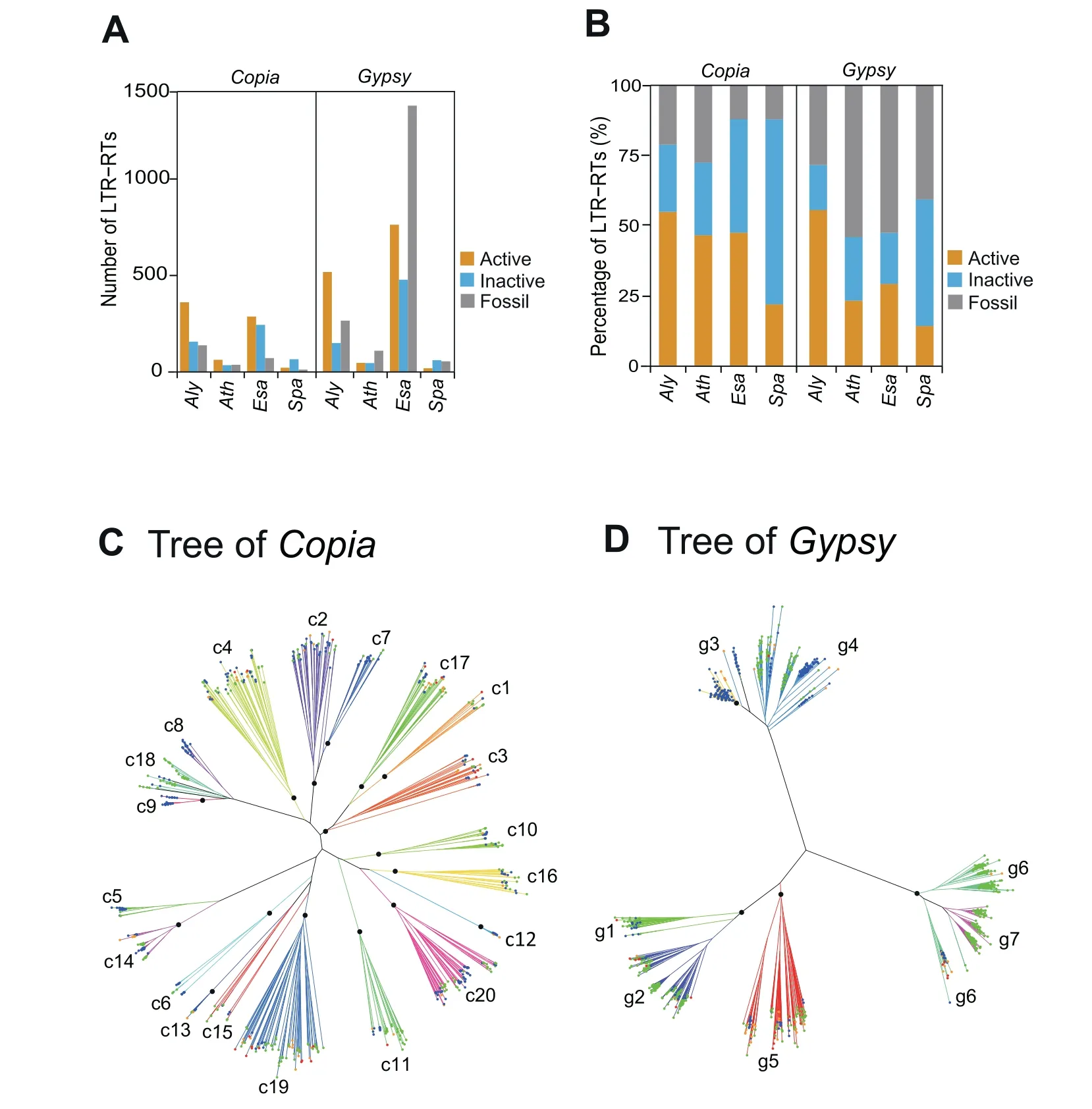
Figure 4 Phylogenetic classification of active LTR-RTs in the four species
The average insertion time of all of the actively transposed members in a clade can be used to estimate the age of an LTRRT clade.The standard deviation (SD) value of the insertion ages in a clade may indicate that the proliferation burst of these active members occurs in a short period time or occurs dispersedly over a long period.Interestingly,we found a significant correlation (Pearson’sr=0.725 and 0.728,P=8.647× 10-10and 2.776 × 10-4forCopiaandGypsyelements,respectively)between the SD and mean values of all clade ages(Figure 5B).However,this correlation between SD and mean values was only true for young clades(average age<2 MYs);a linear regression with an intercept fixed at zero indicated that the mean value was nearly equal to the SD value(slope coefficient=0.878 and 0.889,F-test:P=2.20 × 10-16and 2.36 × 10-9forCopiaandGypsyelements,respectively).In contrast,most of the clades older than 2 MYs displayed a disproportionate drop in their SD values relative to the mean values of insertion time,as evidenced by the gradually flattened fitting curve (Figure 5B).The reason for this phenomenon might be that,comparing to young LTR-RTs,the old ones are more likely purged by negative selection,as mutations have accumulated in the pairs of LTR sequences that have a chance to escape from illegitimate recombination.
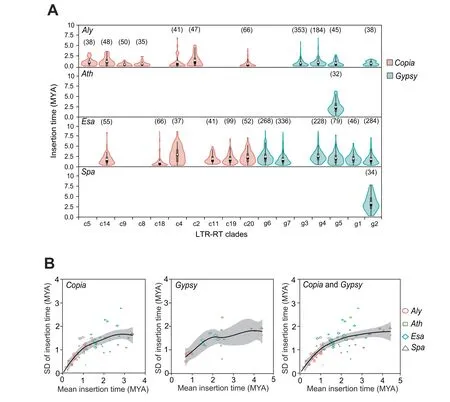
Figure 5 Estimation of the age of the LTR-RT clades in the four species
Species-specific Gypsy proliferation shaped the distinct genome architectures of Aly and Esa
After the RT domain,the INT domain is the second most critical component of functionally active LTR-RTs,because it plays an important role in directing an LTR-RT to integrate into specific locations of the host genome.We observed a more abundant enrichment of activeGypsyelements inEsaandAlycompared toCopiaelements,as well as only specificGypsyclades g5 and g2 enriched inAthandSpa,respectively (Figure 5A).We speculated that the differences in the genomic distribution patterns of theGypsyelements ofEsaandAlymay be attributed to unknown differences in the features of the INT domains of the two species.To test our hypothesis,we first identified activeGypsyelements possessing full-length INT domains with complete coding capacity:61.6%(319/518) inAly,27.7% (13/47) inAth,45.6% (348/763) inEsa,and 10.5% (2/19) inSpa.These ratios were consistent with the contents of youngGypsyinsertions found in the four species.Next,we characterized a specific type of INT domain,the chromodomain(chromatin organization modifier domain),composed of a~50 amino-acid motif that is a signature for chromovirus and can recognize specific genomic locations to be integrated in the host genome [10,14].In plants,chromoviruses are categorised into five clades,the Tekay,CRM,Galadriel,Reina,and Tcn1 clades,which have different genomic integration behaviors [10,28].We collected the sequences of the five known types of chromodomains as templates for the HMMER software package to predict the domains in LTR-RTs in the four species.In total,46 INTs (including 21,8,16 and 1 inAly,Ath,Esa,andSpa,respectively)showed the motif features of chromodomains.Interestingly,with only one exception,the 45Gypsyelements bearing either the Tekay or Reina motif belong to the g5 clade(Figure 6A and B).However,because the C-terminal sequence of the chromodomain is extremely variable,a considerable number of chromodomains might have been overlooked.
In fact,chromoviruses of the CRM clade possess a signature chromodomain motif that has been reported to favour recognition of centromere sequences[5].Gypsyelements carrying the chromodomain motif have a strong bias for insertion into centromeric and/or pericentromeric heterochromatin in plants [14].Thus,we used the HMMER software package to build a HMM profile based on reported CRMGypsyelements in plants to scan the 636 INTs again.The HMM search returned 78 results,including 21 inAly,0 inAth,56 inEsa,and 1 inSpa,showing the signature of a chromodomain motif(Figure 6C).Remarkably,all of theGypsyelements in the results belong to the g2 clade,accounting for 69%of the members in this clade.However,it should be noted that the number of CRMGypsyelements might be underestimated because of the fast-evolving nature of chromodomain motifs.
The genome of the hot pepper also has expanded pericentromeric heterochromatin and is thus four times larger than that of the tomato,a close relative.Kim et al.’s analysis showed that the genome structure of the hot pepper is mainly a result of highly abundantAthila Gypsyelements that preferentially accumulated in heterochromatin [9].Inspired by this work,we collected moreGypsysequences from the GyDB.Using the same HMM prediction procedure,we found that 605 of the 606 members of the g6 and g7 clades,which are specific toEsa,show significant similarity toAthila Gypsyelements,which are completely absent fromAly.In contrast,the g3 clade,which is specific toAly,shows significant similarity toTft2 Gypsyelements belonging to the Tat family,which are mainly present in the euchromatic regions of the hot pepper genome[4,16,29].Therefore,the distinct genomic architectures ofAlyandEsaare highly likely to have been shaped by species-specific clades of activeGypsyelements with distinct INT domains causing preferential integration into euchromatic and heterochromatic regions,respectively.
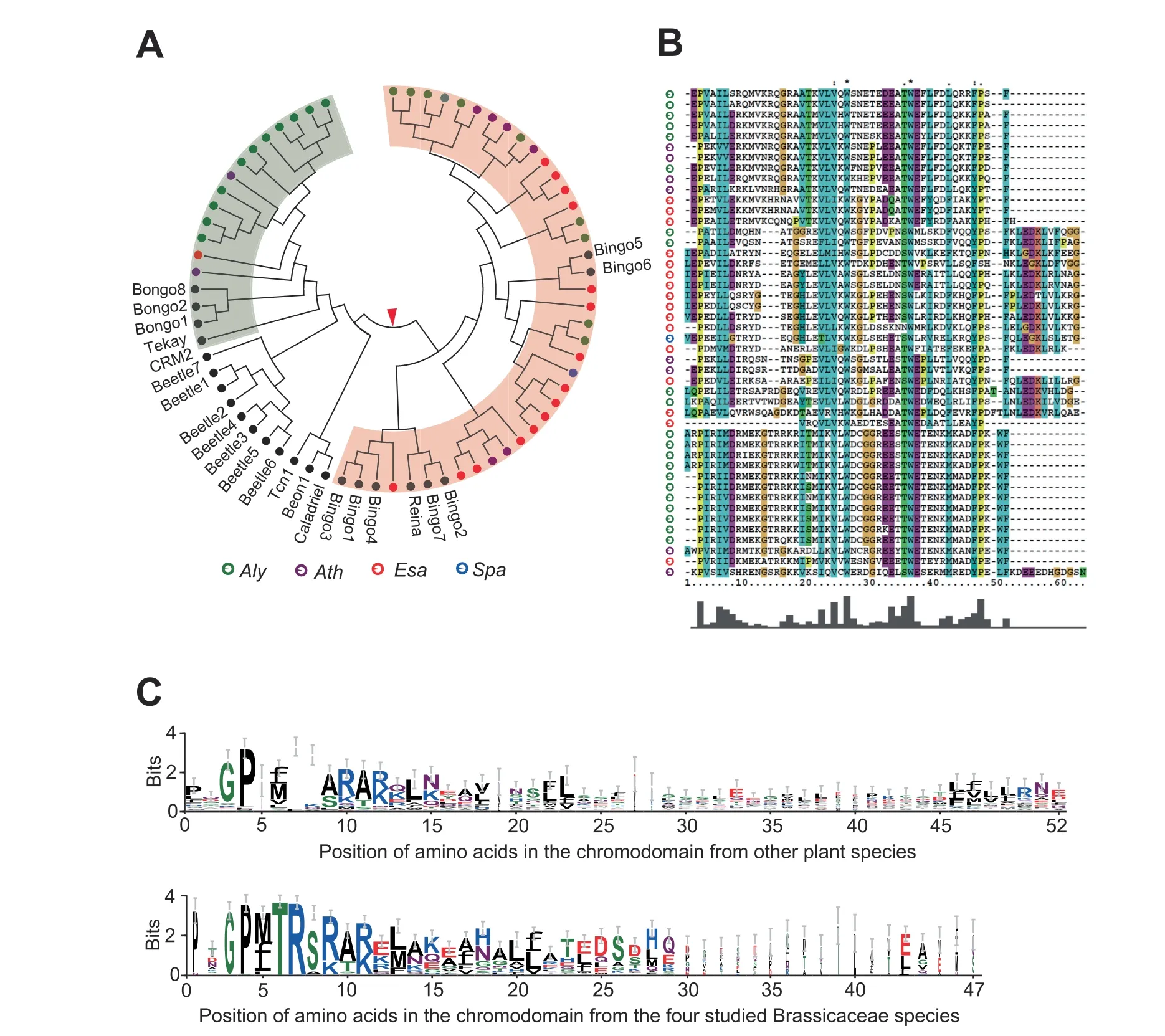
Figure 6 Gypsy elements carrying chromodomains in the four species
Discussion
The activity of Gypsy elements reflects the tendency for genome size evolution
Comparative genomics provides an avenue to infer different evolutionary events during species evolution,such as birth and death of new genes that undergo sub-functionalization and pseudogenization processes,respectively [30].With more and more plant genome sequences are completed,dynamics of genome evolution of different species is far more diverse than what we previously known in the plant kingdom.In addition to frequent polyploidization of higher plants,TE activity is another avenue contributing to the great diversity of plant genome sizes,especially for those closely related species sharing similar evolutionary history.As the coding portions are similar among plant species,the contexts and proportions of TEs,especially retrotransposable elements harbored in intergenic regions,exhibit great diversity.Our comprehensive analyses of LTR-RTs based on a comparison of four Brassicaceae species,in terms of phylogenetic classification,insertion age,transposition bias,and functional domains,indicate the existence of a strong correlation between the tendency for genome size evolution and the activity ofGypsyelements.Nevertheless,it’s worth noting that,because the draft genomes ofAly,Esa,andSpawere assembled based on reads from next-generation sequencing,LTR-RTs might not be completely identified from current versions of the three assemblies.Especially forAlyandEsagenomes enriched withGypsyLTR-RTs,misassembly of repeat-rich sequences may lead to a series of problems when estimating insertion time and performing clade classification.Therefore,our analysis was strictly focused on full-length LTR-RTs with recognizable paired LTRs,in order to minimize possible mistakes due to assembly errors.When new versions of the reference genomes of the three non-model organisms were released,analyses can be redone to validate the conclusion from the current study.
Recent interspecific comparisons in bothOryzaandHelianthusgenera similarly present a strong correlation between the activity of retrotransposable elements and genome size evolution [31,32].Both work indicated that estimated insertion times of LTR-TEs may help infer evolutionary tendency,namely expansion or shrinkage of plant genomes[31,32].Based on the rates of transposition and accumulation of LTR-RTs in the four genomes (Figure 2),and assuming that the rate of DNA loss counteracting TE insertions inAthandEsarepresents a common pace of genome evolution in the Brassicaceae family,it is very likely that the lack of LTR-RT accumulation inSpaover the last two MYs represents a severe depression of TE activity that has facilitated genome shrinkage.In sharp contrast,theAlygenome possesses much more abundant active LTR-RTs with young insertion ages,indicating a recent burst of LTR-RT proliferation within the past 1.5 MYs that led to rapid expansion of its genome.Our work demonstrates that,by analyzing the recent activity of LTR-RTs,particularly the superfamily ofGypsyelements,we may infer the tendency for genome size evolution in plants within a short period of evolutionary time.
Contrasting strategies utilized by two halophytes to reconcile stress-induced TE proliferation
In this work,we selected four fully sequenced species of high relatedness in the Brassicaceae family to investigate the correlation between genome architecture and TEs.WhileEsaandSpaare two natural salt-tolerant halophytic species that inhabit harsh environments that may have high salinity,AthandAlyare salt-sensitive species.Severe abiotic stress can induce activation of LTR-RTs which might be the primary form of long non-coding RNAs functionally involved in stress response [21-33].We initially expected to detect more abundant active LTR-RTs in salt-tolerant speciesEsaandSpathan in salt-sensitive speciesAthandAly.However,we failed to find such a correlation.Despite similar extremophile lifestyles,EsaandSpadisplay dramatic differences in TE content and activity.Nevertheless,this difference may imply thatEsaandSpamight have adopted different molecular mechanisms to reconcile intensive TE transposition under severe environmental stress:Spahas seemingly adopted an active mechanism that rapidly purges deleteriousGypsyinsertions via preferential DNA removal;Esahas seemingly adopted a passive mechanism that confines harmlessGypsyinsertions to gene-poor heterochromatin regions.These distinct strategies of TE defence have resulted in a largeEsagenome that has been subject to intensive centromere expansion,and a smallSpagenome that has been subject to rapid genome downsizing.
Effect of purifying selection on Gypsy insertion in Spa
Genome-wide depletion of theGypsyelements inSpamight be due to efficient DNA removal by innate molecular mechanisms or elimination of individuals carryingGypsy-related sequences from the population by purifying selection.By comparing the ratios of solo-LTRsversuspaired-LTRs ofCopiaandGypsyelements among the four species,we provide evidence that a more efficient DNA removal mechanism mediated by illegitimate recombination may contribute to rapid elimination ofGypsyelements than it does toCopiaelements,indicating the existence of an intrinsic molecular machinery monitoring and controllingGypsyactivities.However,the disproportionate composition and LTR-to-gene distances ofGypsyandCopiaelements suggest that strong purifying selection may also play a role in eliminatingGypsyelements.A fundamental question is whyGypsyelements are more likely to be recognized by the innate DNA removal mechanism if bothGypsyandCopiaelements possess paired identical,direct repeats.One possible explanation for this difference is thatGypsyinsertions might be more toxic thanCopiainsertions to the host genome,and thus subject to strong purifying selection.Two lines of evidence may support this assumption.First,Copia-to-gene distance is much shorter thanGypsy-to-gene distance,suggesting thatCopiainsertion might be less deleterious thanGypsyinsertion,even ifCopiaelements are inserted very close to genes.Second,no significant correlation betweenCopia-togene distance and insertion age was observed inAth,suggesting thatCopiainsertions close to genes might be less harmful thanGypsyinsertions,asGypsyinsertions that occurred close to genes might have been removed by purifying selection.Therefore,the genome contraction process ofSpa,which carries fewerGypsyelements thanEsa,might have been accelerated by strong selection necessitated by the requirement for greater fitness in a harsh environment.Another interesting finding from the analysis ofSpais that a group ofGypsyelements that successfully escaped either DNA removal or purifying selection were primarily young,genic insertions(less than 2 MYs) in protein-coding genes.These insertions may be neutral,less harmful,or even beneficial to genomic adaption to a stressful environment,as TE activity has been hypothesized to be a rapid method of creating raw genetic substrate for natural selection [34].
The transposition preference of specific Gypsy clades leads to centromere expansion in Esa
Studies on multiple plant genomes showed significant enrichment of TEs in pericentromere and centromere regions,mostly epigenetically silenced by DNA methylation and repressive histone marks [35,36].Comparison of the four Brassicaceae genomes showed thatEsahas a unique feature of expanded centromere and pericentromeric heterochromatin because of preferential transposition into gene-poor regions.The transposition bias ofGypsyelements inEsais likely due to an innate mechanism of targeted integration,as reported previously forAth[4,37].This mechanism was inferred from the approximately one-order greaterGypsy-to-gene distances of young insertions (<1 MYs) inEsain comparison with those inAly,as selection pressure may not have had an effect within a short period of evolutionary time [4].However,although the majority ofGypsyinsertions are also distal from genes inAth,this characteristic is likely a result of more efficient elimination of individuals carryingGypsyinsertions close to genes than those carryingGypsyinsertions distal from genes under purifying selection;as apparent from the fitted trend line,the older aGypsyelement,the more likely is it to lie far from genes.Thus,our results provide evidence that natural selection has preferentially purged TEs located near genes more efficiently inAth.Compared toAthandAly,another unique feature ofEsais lacking ofCHROMOMETHYLASE 3(CMT3)gene responsible for gene body methylation,according to a recent report inEsamethylome analysis [38].However,whether missingCMT3has a connection to hugely expanded pericentromere inEsarequires further study.
Another intriguing question is why centromere expansion was not observed inAly,although theAlygenome possesses more activeGypsyelements with young age than does that ofEsa.We reasoned that the distinct architectures of the two genomes are not a result of natural selection,but are instead most likely due to specific clades ofGypsyelements which contain different types of INT domains with different,preferential sites for transpositions.The g6 and g7 clades specifically found inEsa,which account for over 50% of the LTR-RTs inEsa,showed high homology to theAthilafamily,which has been previously reported to preferentially insert into heterochromatin [29].In contrast,the g3 clade specific toAlyshowed significant similarity to theTatfamily which have been reported to be mainly harbored in the euchromatic regions[9].
Materials and methods
Identification of repeat sequences by RepeatMasker
To provide an unbiased estimate of the number of repetitive elements in the four species,RepeatModeler was first applied to constructde novorepeat libraries for the genomes ofAth,Esa,Aly,andSpa,respectively.The fourde novorepeat libraries were then combined with theAthrepeat library,which was downloaded from RepBase.RepeatMasker was used to classify the repeats in the four species based on the combined library.RepeatMasker,RepBase and RepeatModeler were all obtained from (http://www.repeatmasker.org).
Identification of full-length LTR-RTs and solo-LTRs and estimation of insertion time
Full-length LTR-RTs with paired,near-identical,long terminal repeats were predicted using LTR-finder.Because LTRRTs can frequently insert with each other,forming nested patterns,we only used the innermost LTR-RTs to infer insertion times.The insertion times of LTR-RTs were estimated using the Kimura two-parameter distance of paired LTR segments[39].Specifically,the LTR pairs were aligned by MUSCLE(https://www.ebi.ac.uk/Tools/msa/muscle/),and the evolutionary distances were computed by the DISTMAT program implemented in the EMBOSS package (http://emboss.sourceforge.net/).Evolutionary distances were converted into insertion times,assuming an equal neutral substitution rate of 7.0×10-9per site per generation[40].All LTR segments from the full-length LTR-RTs were used as queries to blast(e-value:1.0× 10-6) against the genome sequences to identify homologous fragments.Next,we screened for solo-LTRs that did not overlap with any full-length LTR-RTs.
Categorization of active/inactive/fossil LTR-RTs
We first collected candidate reverse transcriptase (RT)sequences from multiple databases,followed by validation of the structure and completeness of the domain using SMART(http://smart.embl-heidelberg.de/).Then,we selected representative RT domain sequences ofGypsyandCopiaelements to identify the RT regions of the detected full-length LTR-TEs by BLASTX search (e-value:0.1).Finally,each RT domain was manually inspected for any signature that may cause loss of function,such as premature stop codons,frame-shifts,and large insertions and deletions.The LTR-RTs showing complete coding capacity were denoted as‘‘a(chǎn)ctive”;those showing loss-of-function signatures were denoted as ‘‘inactive”;the remaining LTR-RTs,which were severely fragmented,were denoted as ‘‘fossil”.
Classification of active LTR-RTs into clades
The active LTR-RTs were clustered into clades based on a matrix derived from pairwise alignments of RT domain sequences.To measure the similarities among LTR-RTs,we first conducted all-to-all alignments of the LTR-RTs using theneedleallcommand in the EMBOSS package.Then,for each pair of LTRs,within which one LTR at the 5′end of the element was denoted‘‘5′”and the other‘‘3′”,we calculated the normalized similarity score as S5′,3′/min(S5′,5′,S3′,3′),where S5′,3′,S5′,5′,and S3′,3′are the raw scores corresponding to sequence pairs 5′-LTRversus3′-LTR,5′-LTRversus5′-LTR,and 3′-LTRversus3′-LTR,whereas min(S5′,5′,S3′,3′)denotes the minimal value of S5′,5′and S3′,3′.Finally,we performed unsupervised clustering based on the score matrices(M) using the Affinity Propagation (AP) clustering algorithm with theapclusterfunction (negDistMat(r=2),M,details=TRUE) of the apcluster package in R.
Phylogenetic tree construction
The amino acids of RT domains extracted fromGypsyandCopiaelements were aligned by the MUSCLE program.The output files were imported into CLUSTALW to produce the neighbor-joining (NJ) trees.We ran RAxML to build a set of phylogenetic trees under a JTT+gamma model,with 100 rapid bootstraps employed to assess the branch reliability of the NJ tree.
CRediT author statement
Shi-Jian Zhang:Formal analysis,Writing -review &editing.Lei Liu:Resources,Writing -review &editing.Ruolin Yang:Conceptualization,Methodology,Writing -original draft,Writing -review &editing.Xiangfeng Wang:Conceptualization,Methodology,Writing -original draft,Writing -review&editing,Supervision.All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the ‘‘Innovation Project for the Postdoctoral Researchers”in China,and the ‘‘Open Research Fund Program”of Beijing Key Lab of Plant Resource Research and Development,Beijing Technology and Business University.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.07.009.
ORCID
0000-0003-4474-4709 (Shi-Jian Zhang)
0000-0003-3943-6381 (Lei Liu)
0000-0001-6241-6317 (Ruolin Yang)
0000-0002-6406-5597 (Xiangfeng Wang)
 Genomics,Proteomics & Bioinformatics2020年3期
Genomics,Proteomics & Bioinformatics2020年3期
- Genomics,Proteomics & Bioinformatics的其它文章
- CircPlant:An Integrated Tool for circRNA Detection and Functional Prediction in Plants
- Genome Assembly and Pathway Analysis of Edible Mushroom Agrocybe cylindracea
- Paleo-polyploidization in Lycophytes
- Ubiquitinome Profiling Reveals the Landscape of Ubiquitination Regulation in Rice Young Panicles
- Characterization of Lysine Monomethylome and Methyltransferase in Model Cyanobacterium Synechocystis sp.PCC 6803
- Na2CO3-responsive Photosynthetic and ROS Scavenging Mechanisms in Chloroplasts of Alkaligrass Revealed by Phosphoproteomics