
Lipocalin-2 Test in Distinguishing Acute Lung Injury Cases from Septic Mice Without Acute Lung Injury△
2014-04-20 01:39:48GaoZengCongweiJiaJieLiuandShubinGuo
Chinese Medical Sciences Journal 2014年2期
Gao Zeng, Cong-wei Jia, Jie Liu, and Shu-bin Guo*
1Department of Emergency, 2 Department of Pathology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, China
ACUTE lung injury (ALI), or acute respiratory distress syndrome, has been a challenge for emergency physicians for decades. Absence of appropriate biomarkers predicting the onset, progression, and severity of ALI has been hindering the development of an effective pharmacotherapy for this syndrome. In the past decade, a long list of candidates were reported as the potential biomarkers of ALI, such as long pentraxin 3,1,2pre–B-cell colony-enhancing factor,3and the soluble form of the receptor for advanced glycation end products.4Unfortunately, none of these comes into clinical practice because of unsatisfactory sensitivity and specificity, as well as low accessibility. Many proposed tissue damage biomarkers have yet to fulfill the most basic biomarker characteristic that their concentration in the biofluid is proportional to their expression at the injured organ in vivo.5
Lipocalin-2, also known as neutrophil gelatinase associated lipocalin (NGAL) in human, is constitutively expressed in myelocytes and stored in specific granules of neutrophils. It is produced at a high level in a variety of epithelial cells during inflammation, which partly depends on the activation of Toll like receptor 4 (TLR4)-NF-?B signaling pathway.6,7In inflammatory diseases including ulcerative colitis8,9and chronic obstructive pulmonary disease,10lipocalin-2 concentration in the biofluid is associated with the extent of neutrophil infiltration at inflammatory sites. In an animal study on inflammatory response to ischemia and reperfusion in the transplanted heart, lipocalin-2 gene-deficient mice showed dramatically reduced recruitment of granulocytes to the site of ischemia and reperfusion compared to their wild type littermates.11Additionally, lipocalin-2 expression was reported to be prominently up-regulated by reactive oxygen species both in vitro and in vivo, indicating its potential as a surrogate measure for oxidative stress.12Several studies based on murine ALI models induced by pulmonary sterile inflammation reported that pulmonary lipocalin-2 expression was strongly up-regulated by exposure to pro-inflammatory stimuli, suggesting that lipocalin-2 plays an important role in the pathogenesis of inflammatory lung injury.13,14In addition, it has been reported that lipocalin-2 plays a functional role in the regulation of cell apoptosis,15-17and cell apoptosis has been regarded as a key event in the development of sepsis-induced ALI.18,19As a pathogenic component of sepsis and a classical exogenous ligand of pattern-recognition receptor, lipopolysaccharide (LPS) incited many organs including lung to produce a large quantity of lipocalin-2 in dose-dependent manner.5,20A large multicenter prospective study found that plasma NGAL alone was fairly accurate in discriminating septic patients with multi-organ dysfunction from those without.21Taking into account its small size, nature of secretion and relative stability,22we hypothesized that in inflammatory lung injury, the amount of lipocalin-2 in the biofluid was closely related to and regulated by the process of inflammation and could be a potential biomarker of sepsis-induced ALI.
In the current study, we quantified the expression of lipocalin-2 in septic mice, aiming to evaluate the effe- ctiveness of lipocalin-2 test in detecting sepsis-induced ALI.
MATERIALS AND METHODS
Animals
Male C57BL/6 (B6) mice (Vitalriver, Beijing, China) weighing 28-32 g were housed in the animal facility at Peking Union Medical College Hospital, with a controlled environment at a temperature of 22-26°C and a relative humidity of 40%-60% under a 12-hour dark/light cycle. They were fed with a standard laboratory diet and water ad libitum, and were acclimatized for at least 1 week before the experiment. The experiment was carried out in accordance with National Institute of Health guidelines for care and use of laboratory animals and the protocol was approved by the Animal Care and Use Committee of the hospital.
Septic model establishment
Fifty mice were randomly designated by lottery into four sepsis groups (groups A-D) and a non-sepsis group (control group) (n=10 in each group). LPS (Escherichia coli serotype 055:B5, L4524, Sigma, St. Louis, MO, USA) was dissolved in normal saline (1 mg/ml). After intraperitoneal injection of sodium pentobarbital (40-60 mg/kg) for anesthesia, the mice in group A and group B were given LPS (10 mg/kg) by intraperitoneal injection and intravenous injection via tail vein, respectively, and the mice in group C and group D all underwent cecal ligation and puncture (CLP) during which 25% and 75% of the cecum was ligated in group C and group D, respectively. Six mice in the control group were sham-operation controls, which underwent laparotomy under anesthesia with the cecum neither ligated nor perforated; the other 4 mice were saline controls, injected with 0.3 ml normal saline via tail vein under anesthesia. CLP was performed following the protocol proposed by Rittirsch et al.23Briefly, a 1.5-cm-long midline incision was made under anesthesia. The cecum was exposed, ligated at designated positions, and punctured twice with a 21-gauge needle.
Sample collection
Pre-warmed (37°C) normal saline was prepared with buprenorphine (Tianjin Pharmaceuticals, Tianjin, China) to reach the final concentration of 0.4 mg/L. Each of the mice in all the groups was intraperitoneally infused with the saline (1.5 ml) soon after the challenges mentioned above and the infusion was repeated every 4 hours during observations. For the mice in group A, group B, and the saline controls, each observation lasted 6 hours; for those in group C, group D and the sham-operation controls, each observation lasted 48 hours. At the end of the observations, artery blood was collected by abdominal aortic puncture and analyzed immediately with an iSTAT blood gas analyzer and CG4+ cartridges (Abbott Laboratories, Princeton, NJ, USA). The blood samples were allowed to clot overnight at 4°C and centrifuged for 20 minutes at 2000×g. The serum was then collected and stored at -80°C.
The lungs were lavaged thrice via a 22-gauge catheter inserted into the trachea with 1.0 ml ice cold phosphate buffered saline (PBS) containing 5 mmol/L ethylenediamine tetraacetic acid (EDTA), 28 μg/ml aprotinin, and 1 μg/ml leupeptin. The bronchoalveolar lavage fluid (BALF) was temporarily stored at 4°C for further processing within 24 hours.
Pulmonary vasculature was perfused with 4 ml PBS containing 5 mmol/L EDTA at 5 mmHg. The right main bronchus was then ligated and three lobes of the right lung were excised and dried in an oven at 60°C for 72 hours to obtain pulmonary wet-to-dry weight ratio (W/D). One lobe was snap frozen in liquid nitrogen and stored at -80°C for real-time polymerase chain reaction (PCR). The left lung was insufflated with 10% buffered formalin at 25 cm water pressure for 5 minutes, harvested and fixed in 10% buffered formalin for 3 days, and then embedded in paraffin for histopathological examination.
Observation of 7-day survival
Forty-two B6 mice were divided into 5 groups. Septic models were established in group E (n=6), F (n=6), G (n=12), and H (n=12) the same as in group A-D, respectively. Mice in group I (n=6) were sham-operation controls as those in the control group. All the mice received the same fluid treatment as described above during the initial 72 hours of the 7-day observation. They were monitored every 2-8 hours and sacrificed by cervical dislocation when moribund.
Total protein assay and differential cell count in BALF
Besides pulmonary W/D ratios, total protein concentration in BALF was quantified as another measurement of alteration of the alveolar capillary barrier.24After centrifuged for 20 minutes at 1500×g at 4°C, the supernatant of BALF was collected and detected for total protein concentration by a modified Lowry colorimetric assay using the DC protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Along with concentrations of proinflam- matory cytokines and myeloperoxidase protein in the lung, absolute number of neutrophils in BALF was taken as one of the indices of pulmonary inflammatory response. The cell pellet was resuspended in 0.3 ml PBS for cell counting using a standard hemocytometer. A total of 300 cells in each slide were counted for differential cell count using a Diff-Quick- stained kit (Baxter Diagnostics, McGaw Park, IL, USA).
Enzyme-linked immunosorbent assay
Myeloperoxidase in BALF was quantified with mouse myeloperoxidase enzyme-linked immunosorbent assay (ELISA) kits (Hycult Biotech, Uden, The Netherlands). Specific Quantikine ELISA kits (R&D Systems, Minneapolis, MN, USA) were used to quantify mouse interleukin (IL)-6 and lipocalin-2 in BALF and serum. IL-6 was chosen as a representative of proinflammatory cytokines in this study because it was frequently reported increased during the course of sepsis and correlated with the severity and prognosis of this deadly disease.25-27In addition, it was not suggested in consensus by American Thoracic Society that more than one proinflammatory cytokines be tested to define enhanced local inflammatory response.24Each measurement was performed in duplicate according to the manufacturers’ instructions. Concentrations of the proteins were determined by optical densitometry at 450 nm with an automated plate reader (Bio-Rad Laboratories).
Western blotting
Lung tissue samples were homogenized to extract total protein with tissue protein extraction kit (CWBIO, Beijing, China) according to the manufacturer’s instructions. An equal amount (40 μg) of total protein from each sample was separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes (CWBIO), which were blocked with 5% skim milk and sequentially incubated with primary antibodies [rabbit polyclonal anti-mouse lipocalin-2 antibody (Sino Biological, Beijing, China), rabbit polyclonal anti-β-actin antibody (CWBIO), and goat polyclonal anti-mouse IL-6 antibody (R&D Systems)]. The secondary antibodies were horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:2000 dilution) (CWBIO) and HRP-conjugated donkey anti-goat IgG (1:1000 dilution) (R&D Systems). The chemiluminescence detection was performed using the cECL Western blot kit (CWBIO). Films were developed following the standard photographic procedure and quantitative analysis of detected bands was carried out by densitometer scanning using VersaDoc Imaging System (Bio-Rad Laboratories).
Real-time PCR
The snap frozen lung tissue samples were ground in liquid nitrogen and homogenized to extract total RNA using TRIzol Reagent (Life Technologies, Carlsbad, CA, USA). The purity and concentration of the extracted RNA was determined using a spectrophotometer at 260 and 280 nm. The extracted RNA was converted into cDNA with High-Capacity cDNA Reverse Transcription kit (Life Technologies) and real-time PCR was performed in triplicates in a total volume of 50 μl for each run using power SYBR Green PCR master mix on an ABI PRISM 7300 Real-Time PCR System in 96-well PCR plates (Applied Biosystems, Foster City, CA, USA). Specific primer pairs were chemically synthesized (TIANGEN Biotechnology, Beijing, China) and sequences of the primer pairs were: IL-6, 5’-AGTTGCCTTCTTGGGACTGA-3’ (forward) and 5’-TCCACGATTTCCCAGAGAAC-3’ (reverse); lipocalin-2, 5’-AT GTCACCTCCATCCTGGTC-3’ (forward) and 5’-CACACTCACC ACCCATTCAG-3’ (reverse); GAPDH, 5’-TGGGCTACACTGAG CACCAG-3’ (forward) and 5’-GGGTGTCGCTGTTGAAGTCA-3’ (reverse). The initial stage of real-time PCR consisted of 2 minutes at 50°C and 10 minutes at 95°C, followed by 40 cycles of 15 seconds at 95°C plus 1 minute at 60°C. For normalization, expression of gapdh gene was determined in parallel as an endogenous control. Relative gene expression was calculated using the 2-ΔΔCTmethod and expressed as fold change over the expression level in the control group. The PCR products were analyzed by generating melt curves to monitor the presence of nonspecific amplification.
Histopathological examination
Lung tissue sections (4 μm) embedded in paraffin were stained with hematoxylin and eosin (HE). For each sample, 4 successive sections with 5 randomly selected high-power fields (×400) on each slide were analyzed by a pathologist blind to the grouping information. Five histological alterations were considered relevant to the presence of experimental ALI (Table 1). The presence of ≥3 histological alterations in ≥5 high power fields were recorded as the histological evidence for the presence of ALI.
Identification of experimental ALI
The presence of experimental ALI in mice was defined based on 4 categories of measurements (Table 1). The abnormalities in these measurements constituted 4 ‘‘main features’’ of which at least 3 were required to validate the presence of ALI for one condition.24

Table 1. Four categories of evidence for the presence of experimental acute lung injury (ALI)
Diagnostic abilities of lipocalin-2 and IL-6 tests
In order to evaluate the diagnostic abilities of lipocalin-2 and IL-6 tests in BALF and serum in detecting sepsis-induced ALI, a receiver operating characteristic curve (ROC) was generated for each test, and the area under curve (AUC) calculated and compared. The larger the AUC, the better the performance.
Statistical analysis
Statistical analyses were performed using Prism 5.0 (GraphPad Software Inc, USA). Data in normal distribution were expressed as means±SD, and data in skewed distribution expressed as median (interquartile range). Comparisons were carried out using analysis of variance test with the use of Tukey’s multiple comparison test (parametric), or using Kruskal-Wallis test with Dunn’s procedure for post hoc multi-comparison analysis (nonpa- rametric) when data were not assumed sampled from a population with Gaussian distribution. Spearman correlation was calculated among target protein concentrations in the biofluids. Survival curves were derived by the Kaplan-Meier method. P<0.05 was considered statistically significant.
RESULTS
Experimental ALI induced by LPS injection and CLP in mice
In group A and B, LPS challenge resulted in piloerection, huddling, and lethargy by 6 hours. In group C and D, these manifestations became evident on the second post-CLP day. All the control animals kept alert and behaved normally during the experiment.
The total protein in BALF and lung W/D increased significantly in group B and D compared with group A, C and control (all P<0.05, Fig. 1A-B). Myeloperoxidase concentration in BALF, neutrophil count in BALF, and IL-6 level rose significantly in group A, B and D compared with the control group (all P<0.05, Fig. 1C-E), which was accompanied by significantly boosted concentration of serum IL-6 (all P<0.0001, Fig. 1F). Hypoxemia was universal in group B and D, in contrast to no hypoxemia case in group A, C, and the control group. Besides, hyperlactatemia was evident in group B and D (both P<0.0001 compared with the control group, Table 2).
Histological evidence for the presence of ALI were confirmed in 5 cases in group A, 7 cases in group B, 8 cases in group D, 1 case each in group C and the control group. Five mice in group B and D were diagnosed as ALI without histological evidence because they demonstrated the other 3 “main features” of ALI. The mice in group A and C commonly lacked evidence for gas exchange dysfunction and alveolar capillary barrier disruption. The typical histological findings in both group B and group D were substantial accumulation of neutrophils in the lung parenchyma and marked thickening of the alveolar walls with pink staining deposits (Fig. 2B, D), while in group A and group C the characteristic findings were the thin alveolar walls without pink staining deposits and occasional neutrophil infiltrations in the interstitial space (Fig. 2A, C). All the mice in group B and D presented enough evidence to confirm the presence of ALI, while none in the other 3 groups possessed more than 2 of the “main features” of experimental ALI.
Lipocalin-2 and IL-6 levels in the lung and blood
IL-6 in either serum or BALF did not show any statistically significant differences between septic mice with ALI (group B+D) and those without ALI (group A+C) (Fig. 3A, C), while significant increase of lipocalin-2 was revealed both in serum and BALF in septic mice with ALI (both P<0.01, Fig. 3B, D). Semi-quantitative analysis of Western blotting verified significant increases of both proteins of lipocalin-2 and IL-6 in lung tissue in groups A-D compared with the control group (all P<0.0001), while no significant differences were discovered among the 4 sepsis groups (Fig. 4 A-C). Compared with the control group, the transcriptional levels of both genes of lipocalin-2 and IL-6 were significantly increased in the sepsis groups, with even higher levels in group B and group D (Fig. 4D, E). Compared with septic mice without ALI (group A and group C), mRNA expressions of lipocalin-2 (all P<0.05) and IL-6 (all P<0.0001) in lung tissues in septic mice with ALI (group B and group D) were significantly higher (Fig. 4D, E).
Recognition of ALI cases in septic mice by lipocalin-2 test
By comparing the cumulative frequency distribution of data between septic mice with and without ALI, overlapping percentages of the data of IL-6 were found higher than those of lipocalin-2 (80% versus 50% in BALF; 90% versus 45% in serum) (Fig. 5).
Among the tests for both lipocali-2 and IL-6 proteins in serum and BALF, BALF lipocalin-2 test presented the largest AUC in distinguishing ALI cases among septic mice, and serum lipocalin-2 test the second largest (Fig. 6). BALF lipocalin-2 test showed the highest positive likelihood ratio (13.000) at the cutoff value of 1409 ng/ml and the lowest negative likelihood ratio (0.077) at the cutoff value of 773 ng/ml, and serum lipocalin-2 tests performed nearly as well as BALF lipocalin-2 tests (Table 3). A good linear correlation was identified between lipocalin-2 level in BALF and that in serum (Spearman r=0.8803, P<0.0001) (Fig. 7A). Positive correlations were also statistically significant between lipocalin-2 and IL-6 levels in serum (Spearman r=0.7600, P<0.0001) and in BALF (Spearman r=0.5677, P<0.0001) (Fig. 7B, C).
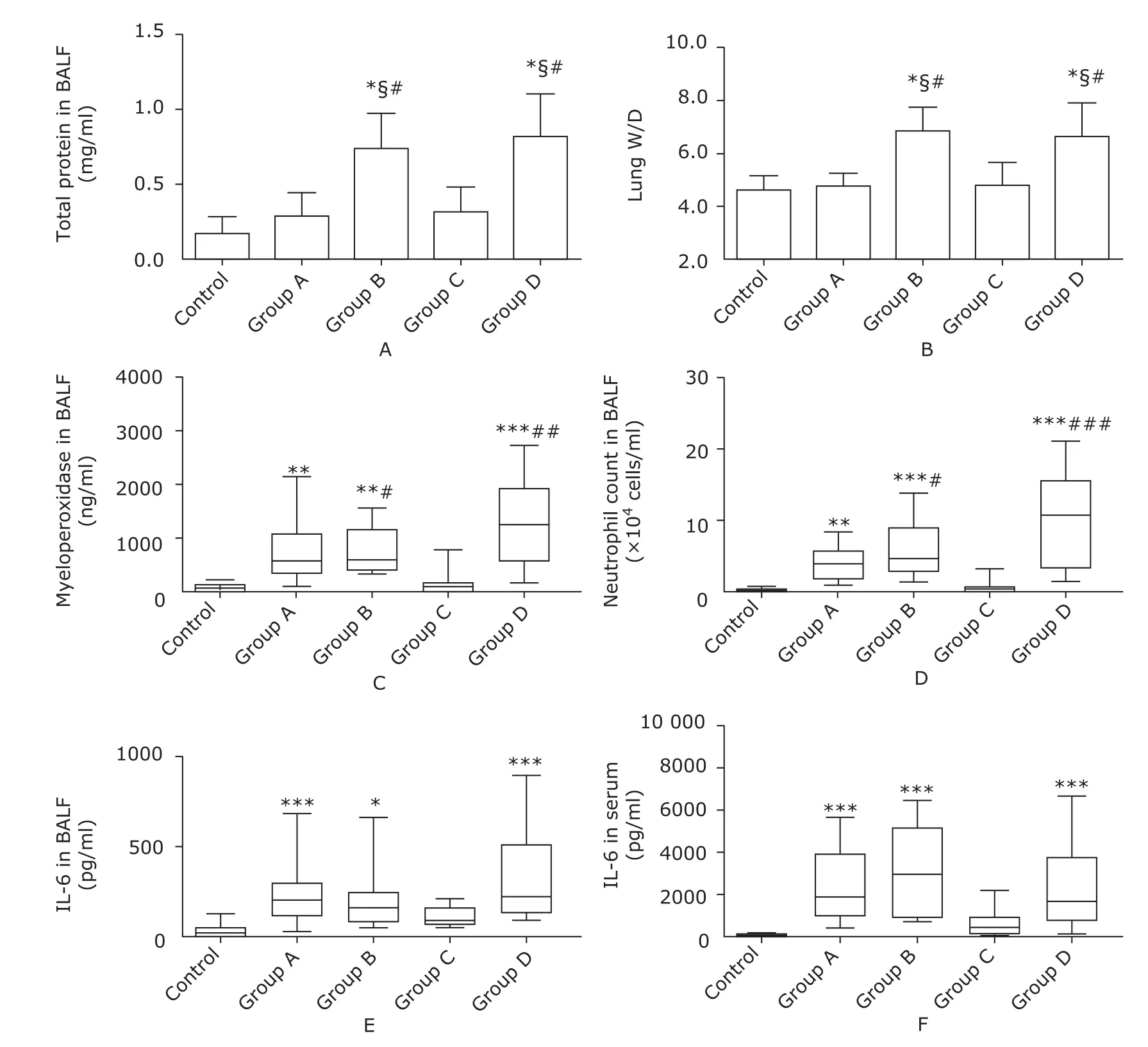
Figure 1. Measurements of alveolar capillary barrier function and inflammatory response. Total protein in BALF (A) and lung W/D (B) were used to measure alveolar-capillary permeability. Myeloperoxidase concentration (C), neutrophil count (D), and IL-6 concentration in BALF (E) were measured to indicate pulmonary inflammatory response. Serum IL-6 concentration (F) reflected systemic inflammatory response. Values were expressed as means±SD (A-B) or median with interquartile range (C-F). The mice in group A (n=10) and group B (n=10) were injected with lipopolysaccharide (10 mg/kg) intraperitoneally and intravenously, respectively. The mice in group C (n=10) and group D (n=10) were treated with cecal ligation and puncture (CLP) ligating 25% and 75% of the cecum, respectively. The control group (n=10) consisted of 6 sham-operation controls and 4 saline controls.*P<0.05, **P<0.01, ***P<0.0001 compared with the control group; §P<0.05 compared with group A; #P<0.05, ##P<0.01, ###P<0.0001 compared with group C.

Table 2. The results of arterial blood gas analysis
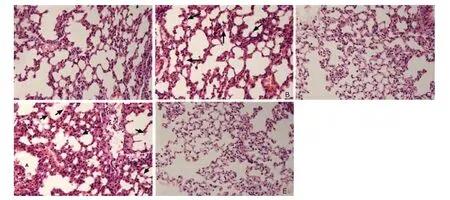
Figure 2. Histological findings of the lung of mice in groups A-D (A-D) and the control group (E) (each n=10). (HE×400) In contrast to the preserved lung parenchymal architecture in the control group, marked thickening of the alveolar walls and pink staining deposits in the alveoli (indicated by arrows) are commonly seen in group B and group D, while in group A and group C the histological findings are characterized by thin alveolar walls and no pink staining deposits.
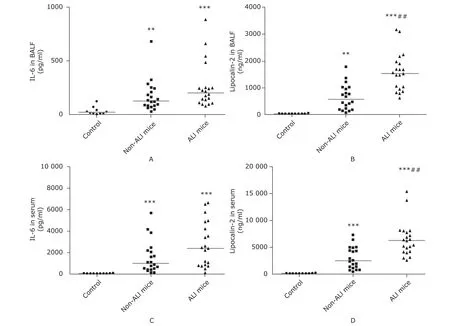
Figure 3. Intergroup comparison of lipocalin-2 and IL-6 in the biofluid. The concentrations of the proteins determined by enzyme-linked immunosorbent assay are expressed as medians with range, which includes IL-6 in BALF (A), lipocalin-2 in BALF (B), IL-6 in serum (C), and lipocalin-2 in serum (D). Non-ALI mice (n=20) were those in group A and C which had developed sepsis but failed to be diagnosed as ALI cases. ALI mice (n=20) were those in group B and D which had developed both sepsis and ALI. **P<0.01, ***P<0.0001 compared with the control group; ##P<0.01 compared with non-ALI mice.
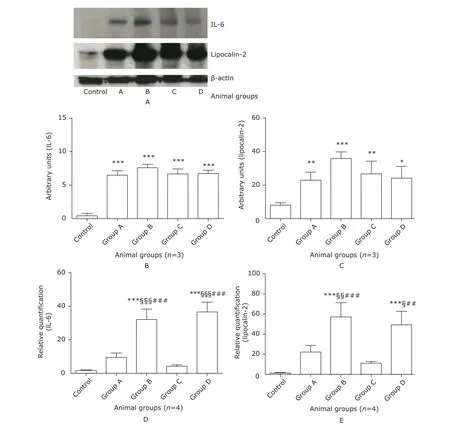
Figure 4. The expression of lipocalin-2 and IL-6 in the lung of mice. Whole tissue lysate of mouse lung was subjected to Western blotting with anti-lipocalin-2, anti-IL-6, and anti-β-actin antibodies. Representative bands were shown in (A). Specific bands of IL-6 and lipocalin-2 were quantified by densitometry and expressed as mean±SD. Significant increases in IL-6 (B) and lipocalin-2 (C) proteins were present in groups A-D compared with the control group. Total RNA in the lung was extracted for real-time PCR, which was performed in triplicates. Expression of gapdh gene was determined as an endogenous control. Relative gene expression of IL-6 (D) and lipocalin-2 (E) was calculated using the 2-Δ Δ CT method and expressed as fold increase over the control group. *P<0.05, **P<0.01, ***P<0.0001 compared with the control group; §P<0.05, §§P<0.01, §§§P<0.0001 compared with group A; ##P<0.01, ###P<0.0001 compared with group C.
Survival rate of septic mice with or without ALI
None of the mice in group H, which were subjected to CLP with the cecum 75% ligated, lived through 84 hours, with 10 deaths occurring on the third post-CLP day. The mice in group G, which were subjected to CLP with the cecum 25% ligated, had only 2 deaths during the 7-day follow-up. The mice in group F receiving intravenous LPS (10 mg/kg) died by 16 hours, and the earliest deaths occurred at about 8 hours after LPS injection. There was no death in the mice in group E receiving intraperitoneal LPS (10 mg/kg) or the mice in group I (the sham-operation controls) in the 7-day follow-up (Fig. 8). By the end of the 7-day follow-up, 81.8% of the survivals (18 out of 22) were witnessed restoring normal exploratory and feeding behavior.
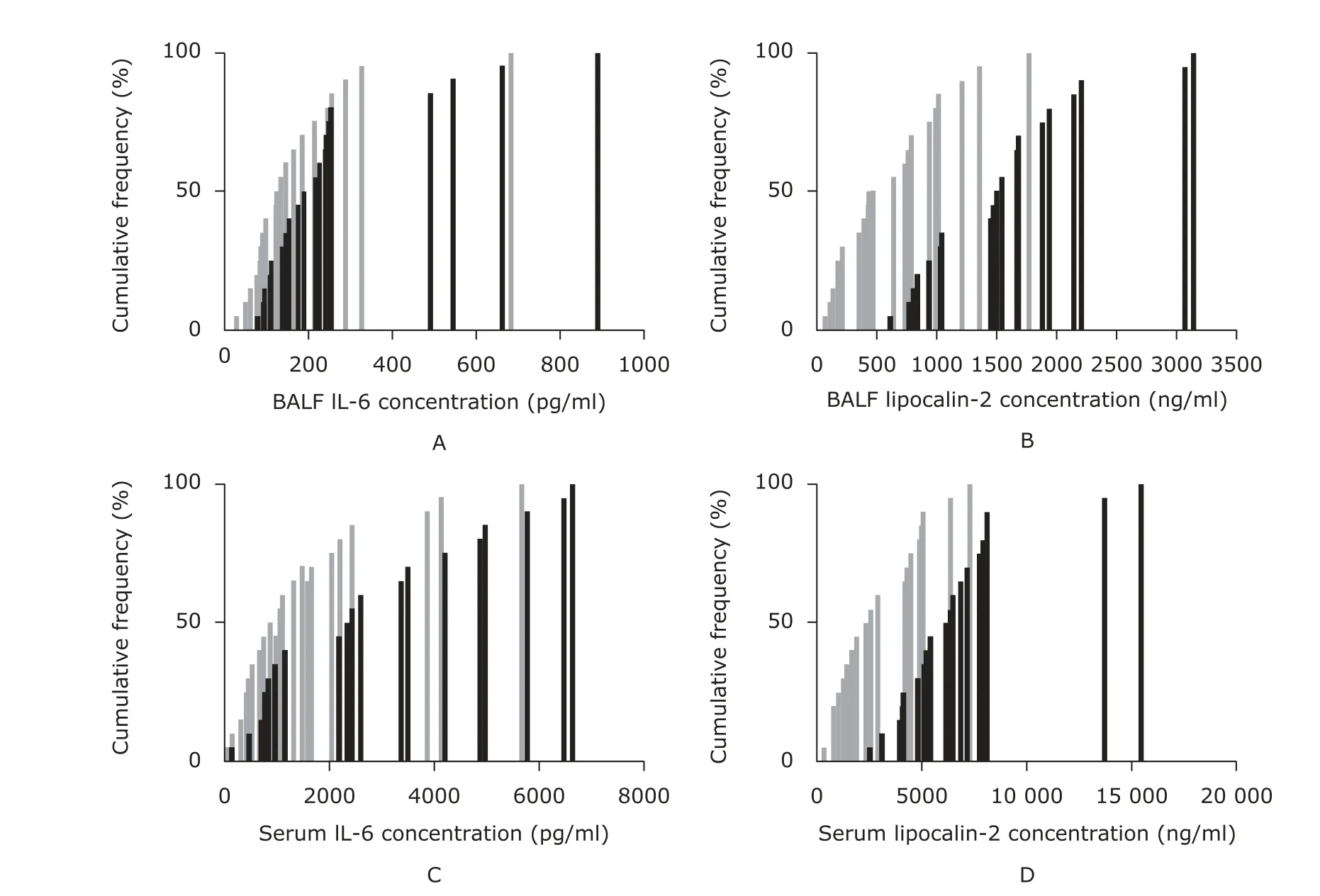
Figure 5. The comparison between ALI mice and non-ALI mice in terms of cumulative frequency distribution of the concentrations of IL-6 and lipocalin-2 in BALF and serum. The concentrations of IL-6 (A, C) and lipocalin-2 (B, D) in BALF and serum were determined by enzyme-linked immunosorbent assay. Non-ALI mice (n=20, indicated by gray color) were those in group A and C. ALI mice (n=20, indicated by black color) were those in group B and D.
DISCUSSION
Sepsis is the most commonly encountered condition underlying the development of ALI. Many animal models have been developed to study the pathogenesis of sepsis- induced ALI. In 2011, a workshop initiated by the American Thoracic Society first reported an expert consensus on the definition of experimental ALI, aiming to help investigators to determine whether they have achieved (or prevented) lung injury in animal models.24We adopted the definition in this study to confirm the presence of ALI in murine models with certain simplification to mitigate inter- and intra-observer variation.
Intravenous infusion of LPS is one of the earliest approaches used to induce ALI in animals, which targets primarily the capillary endothelium.28In our pilot study, this method with LPS dosage ranging between 5-10 mg/kg typically resulted in lung tissue injury within 4-6 hours and caused nearly 100% lethality by 24 hours in murine models. Given adequate volume resuscitation, intraperitoneal infusion of LPS (≤20 mg/kg) rarely caused most of the “main features” of ALI in B6 mice and led to few deaths in 14-day follow-up. CLP is the most widely used method to establish clinically relevant sepsis models, but it is not so often used to model experimental ALI.28The ligated position of the cecum is a major determinant of lethality in peritonitis rodents, and the majority of deaths often emerged between 24-48 hours after CLP.23Recently, Iskander et al29reported that no signs of lung injury were found in murine models of CLP-induced sepsis, which led to a conclusion that “in murine intra-abdominal sepsis, pulmonary injury cannot be considered the etiology of death in the acute phase”. In their study, the ligated position of the cecum was not reported, and mice routinely received antibiotic therapy which undoubtedly contributed to the low 7-day mortality (less than 20%). In the present study, ligation of a large proportion of the cecum and adequate time for pathological development were the keys to reproducing ALI in murine peritonitis models, and active fluid resuscitation helped the mice to survive no less than 48 hours post-CLP. Impaired oxygenation, alveolar capillary barrier dysfunction, and enhanced pulmonary inflammation converged on the mice that were either intravenously injected with LPS (10 mg/kg) or subjected to CLP with 75% of the cecum ligated, which indicated that ALI had developed.

Figure 6. The diagnostic abilities of lipocalin-2 and IL-6 tests to discriminate ALI among septic mice. According to the receiver operating characteristic curve (ROC) and area under curve (AUC), BALF IL-6 test could hardly recognize ALI cases among septic mice (A), but serum IL-6 test performed slightly better (C). Serum lipocalin-2 test (D) performed nearly as well as BALF lipocalin-2 test did (B).
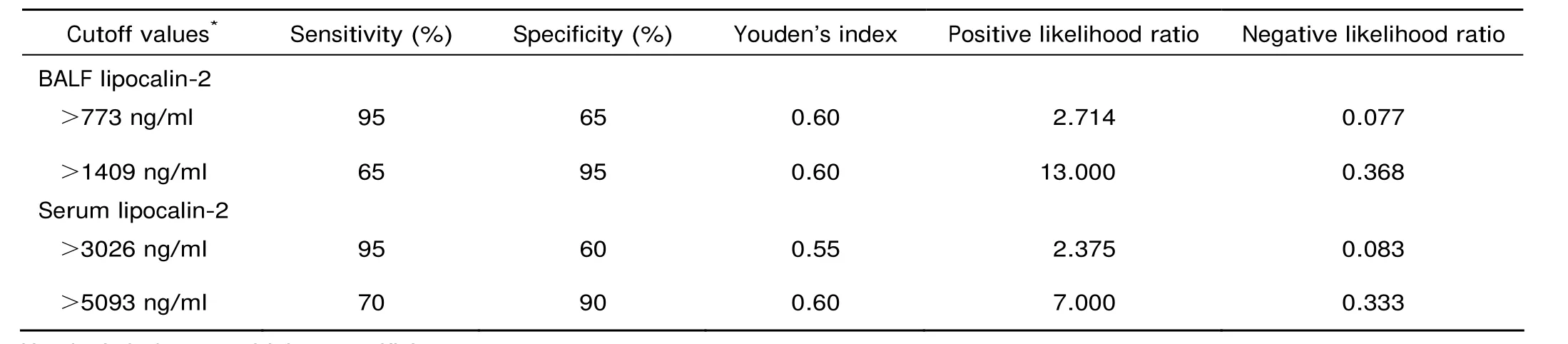
Table 3. Validity evaluation of lipocalin-2 tests with a dual cutoff system
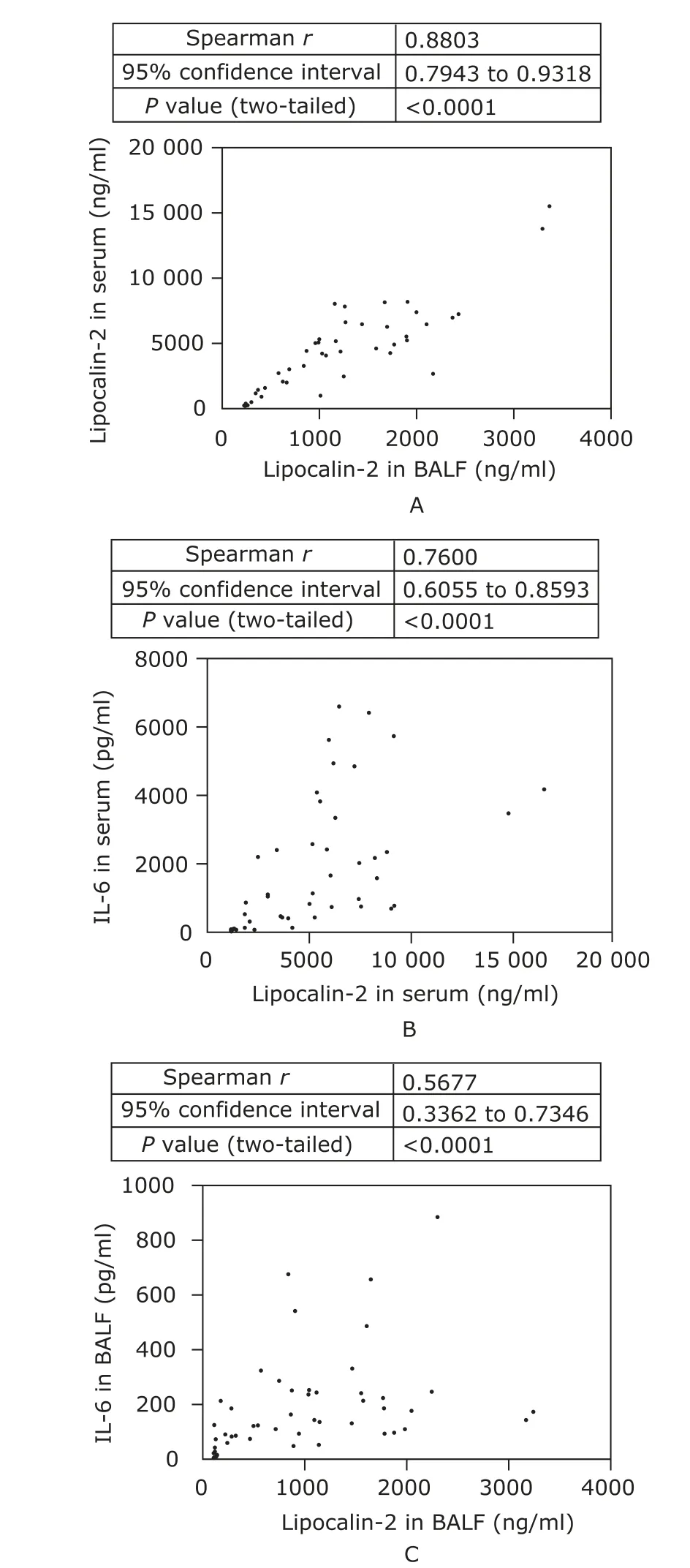
Figure 7. Correlation analysis of the concentrations of two proteins in the biofluids. Lipocalin-2 concentration in serum correlated well with that in BALF (A). Positive correlations were significant between lipocalin-2 and IL-6 concentration both in serum (B) and in BALF (C).
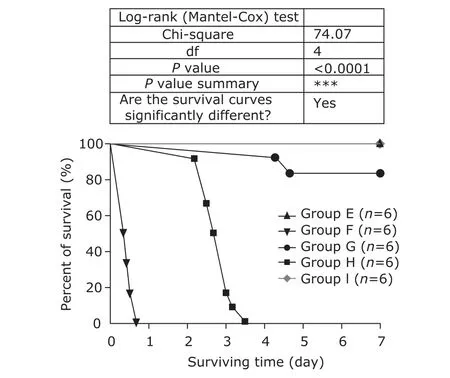
Figure 8. Seven-day survival of septic mice. The mice in group E and group F were challenged with lipopolysaccharide (LPS) (10 mg/kg) by intraperitoneal and intravenous injection, respectively; those in group G and group H underwent the same surgical procedure of cecal ligation and puncture (CLP) with 25% and 75% of the cecum ligated, respectively; those in group I were subjected to laparotomy with the cecum neither ligated nor punctured (sham-operation). The mice were monitored every 2-8 hours and euthanized by cervical dislocation when moribund. The symbols on the survival curve represent the time when death events occurred.
The expression of inflammatory cytokines including IL-6 is known to be up-regulated partly by NF-κB activation, and activated NF-κB has been taken as a positive regulator of lipocalin-2 expression.22IL-6 stimulated over-production of lipocalin-2 in rodent hepatocytes in vitro.30Furthermore, a massive increase in gene expression of lipocalin-2 was ob- served in mouse liver during acute phase reaction, which was significantly inhibited in il-6 knockout mice, indicating IL-6 could be the main cytokine regulating lipocalin-2 production.31In this study, IL-6 levels correlated well with lipocalin-2 levels in the biofluid, supporting an intimate biological relevance between the two proteins. It has been reported that treatment aiming at blocking the biological effects of IL-6 improved the survival of septic mice,32,33suggesting a functional role of IL-6 in the pathogenesis of severe sepsis and multiple organ injury. Taking into account the functional role of lipocalin-2 in the regulatory process of inflammation and cell apoptosis, it is logical to infer that both of the proteins take part in orchestrating sepsis-induced organ injury. The inconsistent statistical difference of the two proteins in the biofluid between septic mice with and without ALI can be partly explained by the following considerations: firstly, IL-6 can be degraded very rapidly by proteases in inflammatory exudate, while lipocalin-2 is well known as protease resistant;22secondly, the intergroup differences of IL-6 in the biofluid could be too small to be detected by a nonparametric calculation, especially in small sample size. It also should be noted that we were not sure why real-time PCR detected significant differences in the expression of both genes between septic mice with and without ALI while Western blotting did not show the similar pattern of intergroup differences at protein level. Perhaps, owing to the rich production in epithelia, secreted nature, and proteases resistance of lipocalin-2,22its intergroup differences may be greater in BALF than in lung tissue lysate during sepsis. In contrast, IL-6 in the biofluids tends to be rapidly degraded by proteinases and exhibits no statistical difference between septic mice with and without ALI. It may also be possible that there actually exists discordance between transcriptional levels and translational levels of both lipocalin-2 and IL-6 genes during sepsis.
In some studies, IL-6 was adopted as an early marker to predict the mortality of experimental sepsis.26,27,34Nevertheless, few studies argued for its utility to mark sepsis-induced organ injury. Similarly, IL-6 tests in this study showed much less power than lipocalin-2 tests to recognize ALI cases among septic mice. This was especially true for BALF IL-6 test, which produced the worst performance in diagnosing ALI. On the contrary, lipocalin-2 tests had a good performance in distinguishing septic ALI mice from those without ALI. Interestingly, lipocalin-2 levels in BALF correlated well with those in serum, fulfilling the most basic requirement for ideal organ injury biomarkers, i.e. their concentration in the biofluid should keep proportional to their expression at the site of injury in vivo. In clinical settings, BALF specimens are not routinely acquired; therefore the tests are not easy to be normalized. On the other hand, blood samples are easily and normally obtained, which makes blood test results more reliable and suitable for comparison among different laboratories. In this regard, serum lipocalin-2 tests have demonstrated a promising ability in the diagnosis of sepsis-induced ALI.
There are some limitations in this study. Due to difficulty in intermittent sampling in small animals and lack of reliable methodology to quantify experimental lung injury, we did not study whether the levels of lipocalin-2 in the biofluid reflected a graded, dose-dependent lung injury. In addition, we did not know the time-course changes of lipocalin-2 in the pathological progression of sepsis-induced ALI. More laboratory efforts are needed to elucidate the details about how lipocalin-2 functions in the pathogenesis of sepsis-induced ALI.
In conclusion, lipocalin-2 expression in both lung and blood was significantly up-regulated in septic mice with ALI compared with those without ALI. Lipocalin-2 tests with a dual cutoff system performed well in distinguishing experimental ALI cases.
1. Mauri T, Coppadoro A, Bellani G, et al. Pentraxin 3 in acute respiratory distress syndrome: an early marker of severity. Crit Care Med 2008; 36:2302-8.
2. He X, Han B, Bai X, et al. PTX3 as a potential biomarker of acute lung injury: supporting evidence from animal experimentation. Intensive Care Med 2010; 36:356-64.
3. Ye SQ, Simon BA, Maloney JP, et al. Pre–B-cell colony- enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med 2005; 171:361-70.
4. Jabaudon M, Futier E, Roszyk L, et al. Soluble form of the receptor for advanced glycation end products is a marker of acute lung injury but not of severe sepsis in critically ill patients. Crit Care Med 2011; 39:480-8.
5. Paragas N, Qiu A, Zhang Q, et al. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 2011; 17:216-22.
6. Borregaard N, Cowland JB. Neutrophil gelatinase-associated lipocalin, a siderophore-binding eukaryotic protein. BioMetals 2006; 19:211-5.
7. Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics 1997; 45:17–23
8. Chassaing B, Srinivasan G, Delgado MA, et al. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One 2012 [cited 2013 Nov 1]; 7: e44328. Availabe from: http://www. plosone.org/article/info%3Adoi%2F10.1371%2Fjournal. pone.0044328.
9. Carlson M, Raab Y, Sevéus L, et al. Human neutrophil lipocalin is a unique marker of neutrophil inflammation in ulcerative colitis and proctitis. Gut 2002; 50:501-6.
10. Keatings VM, Barnes PJ. Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjects. Am J Respir Crit Care Med 1997; 155:449-53.
11. Aigner F, Maier HT, Schwelberger HG, et al. Lipocalin-2 regulates the inflammatory response during ischemia and reperfusion of the transplanted heart. Am J Transplant 2007; 7:779-88.
12. Roudkenar MH, Kuwahara Y, Baba T, et al. Oxidative stress induced lipocalin 2 gene expression: addressing its expression under the harmful conditions. J Radiat Res 2007; 48:39-44.
13. André E, Stoeger T, Takenaka S, et al. Inhalation of ultrafine carbon particles triggers biphasic pro-inflammatory response in the mouse lung. Eur Respir J 2006; 28: 275-85.
14. Connor AJ, Laskin JD, Laskin DL. Ozone-induced lung injury and sterile inflammation. Role of toll-like receptor 4. Exp Mo Pathol 2012; 92:229-35.
15. Devireddy LR, Gazin C, Zhu X, et al. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005; 123:1293-305.
16. Lee S, Lee J, Kim S, et al. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol 2007; 179:3231-41.
17. Xu G, Ahn J, Chang S, et al. Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J Biol Chem 2012; 287:4808-17.
18. Perl M, Lomas-Neira J, Venet F, et al. Pathogenesis of indirect (secondary) acute lung injury. Expert Rev Resp Med 2011; 5:115-26.
19. Perl M, Chung CS, Perl U, et al. Therapeutic accessibility of caspase mediated cell death as a key pathomechanism in indirect acute lung injury. Crit Care Med 2010; 38:1179-86.
20. Sunil VR, Patel KJ, Nilsen-Hamilton M, et al. Acute endotoxemia is associated with upregulation of lipocalin 24p3/Lcn2 in lung and liver. Exp Mo Pathol 2007; 83:177-87.
21. Shapiro NI, Trzeciak S, Hollander JE, et al. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med 2009; 37:96-104.
22. Chakraborty S, Kaur S, Guha S, et al. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta 2012; 1826:129-169.
23. Rittirsch D, Huber-Lang MS, Flierl MA, et al. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 2009; 4:31-6.
24. Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 2011; 44:725-38.
25. Damas P, Ledoux D, Nys M, et al. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann Surg 1992; 215:356-62.
26. Osuchowski MF, Connett J, Welch K, et al. Stratification is the key: inflammatory biomarkers accurately direct immunomodulatory therapy in experimental sepsis. Crit Care Med 2009; 37:1567-73.
27. Remick DG, Bolgos GR, Siddiqui J, et al. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 2002; 17:463-7.
28. Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008; 295:L379-99.
29. Iskander KN, Craciun FL, Stepein DM, et al. Cecal ligation and puncture-induced murine sepsis does not cause lung injury. Crit Care Med 2013; 41:159-70.
30. Sultan S, Pascucci M, Ahmad S, et al. Lipocalin-2 is a major acute-phase protein in a rat and mouse model of sterile abscess. Shock 2012; 37: 191-6.
31. Luchtefeld M, Preuss C, Rühle F, et al. Gp130-dependent release of acute phase proteins is linked to the activation of innate immune signaling pathways. PLoS One 2011 May 4 [cited 2013 Nov 19]; 6: e19427. Available from: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0019427
32. Riedemann NC, Neff TA, Guo RF, et al. Protective effects of IL-6 blockade in sepsis are linked to reduce C5a receptor expression. J Immunol 2003; 170:503-7.
33. Mostafa Anower AKM, Shim JA, Choi B, et al. Pretreatment with interleukin-6 small interfering RNA can improve the survival rate of polymicrobial cecal ligation and puncture mice by down regulating interleukin-6 production. Eur J Pharmacol 2012; 688:76-83.
34. Craciun FL, Schuller ER, Remick DG. Early enhanced local neutrophil recruitment in peritonitis-induced sepsis improves bacterial clearance and survival. J Immunol 2010; 185:6930-8.
 Chinese Medical Sciences Journal2014年2期
Chinese Medical Sciences Journal2014年2期
- Chinese Medical Sciences Journal的其它文章
- Serum Levels of Interleukin-1 Beta, Interleukin-6 and Melatonin over Summer and Winter in Kidney Deficiency Syndrome in Bizheng Rats△
- Minimally Invasive Perventricular Device Closure of Ventricular Septal Defect: a Comparative Study in 80 Patients
- Expression of Peptidylarginine Deiminase 4 and Protein Tyrosine Phosphatase Nonreceptor Type 22 in the Synovium of Collagen-Induced Arthritis Rats△
- False Human Immunodeficiency Virus Test Results Associated with Rheumatoid Factors in Rheumatoid Arthritis△
- Arachnoiditis Ossificans of Lumbosacral Spine: a Case Report and Literature Review
- Ketamine Abuse-Induced Obstructive Nephropathy and Kidney Injury