
Genome-wide association studies reveal the genetic basis of amino acid content variation in tea plants
2023-11-18 09:32:14GUOYafeiLlDailiQlUHaijiZHANGXiaoliangLlULinZHAOJingjingJlANGDeyuan
Journal of Integrative Agriculture 2023年11期
GUO Ya-fei, Ll Dai-li, QlU Hai-ji, ZHANG Xiao-liang, LlU Lin, ZHAO Jing-jing, JlANG De-yuan
National Key Laboratory for Germplasm Innovation & Utilization of Horticultural Crops, College of Horticulture and Forestry Sciences, Huazhong Agricultural University, Wuhan 430070, P.R.China
Abstract Tea is one of the most popular non-alcoholic beverages in the world, and free amino acids, especially theanine, make a major contribution to the umami taste of tea.However, the genetic basis of the variation in amino acid content in tea plants remains largely unknown.Here, we measured the free amino acid content in fresh leaves of 174 tea accessions over two years using a targeted metabolomics approach and obtained genotype data via RNA sequencing.Genomewide association studies were conducted to investigate loci affecting the content of free amino acids.A total of 69 quantitative trait loci (–log10(P-value)>5) were identified.Functional annotation revealed that branched-chain amino acid aminotransferase, glutamine synthetase, nitrate transporter, and glutamate decarboxylase might be important for amino acid metabolism.Two significant loci, glutamine synthetase (Glu1, P=3.71×10–4; Arg1, P=4.61×10–5) and branched-chain amino acid aminotransferase (Val1, P=4.67×10–5; I_Leu1, P=3.56×10–6), were identified, respectively.Based on the genotyping result, two alleles of CsGS (CsGS-L and CsGS-H) and CsBCAT (CsBCAT-L and CsBCAT-H) were selected to perform function verification.Overexpression of CsGS-L and CsGS-H enhanced the contents of glutamate and arginine in transgenic plants, and overexpression of CsBCAT-L and CsBCAT-H promoted the accumulation of valine, isoleucine and leucine.Enzyme activity assay uncovered that SNP1054 is important for CsGS catalyzing glutamate into glutamine.Furthermore, CsGS-L and CsGS-H differentially regulated the accumulation of glutamine, and CsBCAT-L and CsBCAT-H differentially regulated the accumulation of branched-chain amino acids.In summary, the findings in our study would provide new insights into the genetic basis of amino acids contents variation in tea plants and facilitate the identification of elite genes to enhance amino acids content.
Keywords: Camellia sinensis, amino acids, genetic variation, association studies, genotype analysis, functional verification
1.lntroduction
Tea (Camelliasinensis(L.) O.Kuntze) is an important crop, and the young leaves of tea plants possess diverse amino acids such as theanine (Thea), glutamate(Glu), proline (Pro), arginine (Arg) and aspartic acid(Asp).Amino acids are not only important nitrogenous components in tea plants, but they are also the main form of nitrogen transported between different organs through the xylem and phloem (Besnardet al.2016).Therefore,amino acids comprise the nitrogen pool in tea plants.The content of free amino acids in fresh leaves, especially theanine, is closely related to tea flavor (umami) and yield of tea plants (Ruanet al.2010).In general, amino acids are an important indicator for studying tea quality and nitrogen assimilation in tea plants.However, the genetic basis of natural variation in amino acid content in tea plants has not yet been clarified.
In recent years, many genes involved in the amino acid pathways in tea plants have been isolated by reverse genetic studies, such asCsAlaDC(Baiet al.2019),CsAAPs(Donget al.2019),CsPDX2.1(Fuet al.2020),CsLHT1(Liet al.2021),CsTSI(Weiet al.2018),andCsMYB73(Wenet al.2020).Forward genetic approaches, including quantitative trait loci (QTL)mapping and association mapping, are important tools for identifying loci regulating free amino acid content in higher plants.InGlycinemax, Khandakeret al.(2015) identified 16 QTLs associated with seven amino acid traits through QTL mapping.Zhonget al.(2011) measured protein content and amino acid content in an F9 population of recombinant inbred line and identified 112 QTLs in rice by QTL mapping.In addition, Xuet al.(2014) identified 17 QTLs associated with the content of lysine (Lys),threonine (Thr) and methionine (Met) in rapeseed meal by QTL mapping.In tea plants, QTL mapping has been used to identify loci regulating variation in metabolic traits.Four QTLs related to free amino acid content in a segregating population of ‘Longjing 43’בBaijiguan’ were identified(Huanget al.2022).Xuet al.(2018) identified 27 QTLs associated with flavonoids or caffeine by QTL mapping.Maet al.(2018) identified 10 stable QTLs associated with the content of alkaloid (caffeine, theobromine) by QTL mapping in tea plants.
Camelliasinensisis a perennial and self-incompatible woody plant with a large genome, long generation cycle,and high heterozygosity, which makes it building a large-scale segregating population time-consuming and laborious.Unlike QTL mapping, genome-wide association studies (GWASs) are a quick, inexpensive, and highresolution tool for identifying genetic variants in higher plants.Currently, GWASs have been extensively used for the study of metabolite traits in many model plants and widely cultivated crops, such asArabidopsisthaliana(Filiault and Maloof 2012),Oryzasativa(Sunet al.2020),Zeamays(Wenet al.2014),Triticumaestivum(Chenet al.2020),Brassicanapus(Wanget al.2017),Glycine max(Zhanget al.2019), andSolanumlycopersicum(Yeet al.2017).Branched-chain amino acids (BCAAs),including valine (Val), isoleucine (Ile), and leucine (Leu),are essential amino acids in animals and humans because they cannot be synthesized by themselves.InO.sativa, association studies found that natural variation in the promoter region ofOsbZIP18was identified to be associated with the content of BCAAs (Sunet al.2020).InA.thaliana, Angeloviciet al.(2013) identified an allelic variant ofAtBCAT2that was associated with BCAA content.With the rapid development of next-generation sequencing technology, GWASs have been conducted to study metabolic traits in tea plants.Recently, Wanget al.(2019) identified 27 single-nucleotide polymorphisms(SNPs) associated with the timing of spring bud flush in 151 tea plant germplasm resources using GWAS.Fanget al.(2021) performed a GWAS in a natural population of 191 tea accessions and identified 80 SNPs associated with the caffeine content, 7 SNPs associated with the theanine content, and 293 SNPs associated with the content of catechins.Free amino acids are the main component of umami and sweet taste of tea infusion,which can effectively counteract the astringency of catechins and tannins, as well as the bitterness of caffeine and theacrine (Zhang Let al.2020).Nevertheless, few studies have used GWASs to explore the genetic basis of natural variation in the content of free amino acids in tea plants.
Here, we measured the content of free amino acids in the young leaves of 174 tea plants in 2018 and 2019.We conducted a GWAS based on the genotype data of 174 tea accessions obtained by RNA sequencing to identify loci associated with the content of free amino acids.Many loci and candidate genes were identified.Two candidate genes,CsGSandCsBCAT, were subjected to genotype analysis, and their functions were verified.It was found thatCsGSwas involved in the conversion of Glu to glutamine (Gln) and thatCsBCATplays a crucial role in the biosynthesis of Val, Ile, and Leu.In addition,genetic variant in the coding sequences ofCsGSandCsBCATaffects their role in regulating the accumulation of free amino acids in transgenic plants.Overall, the data acquired in this study provide abundant genetic information for the study of key genes involved in amino acid metabolic pathway in tea plants.
2.Materials and methods
2.1.Plant materials
A natural population consisting of 174 individuals was planted in the tea plant nursery of Huazhong Agricultural University, Wuhan, China.The information on the 174 tea accessions was the same as that described in previous study (Zhang W Yet al.2020).Young leaves (one bud with a leaf, YL) from 174 tea accessions were collected in July 2018 (YL1) and April 2019 (YL2).Samples were frozen in liquid nitrogen and stored at –80°C.Samples were then frozen dried and prepared for metabolite extraction.NicotianabenthamianaandA.thaliana(Col-0)were cultured in a greenhouse (14 h/10 h, day/night,23°C).
2.2.Determination of the content of amino acids by UPLC-QQQ-MS
Free amino acids were extracted with an aqueous methanol solution.First, the frozen-dried samples were finely ground to powder.The samples (50 mg) were extracted with 1 mL of 70% methanol solution for 13 h at 4°C.The mixture was centrifuged at 12 000 r min–1for 15 min at 4°C.The supernatant was then filtered through a 0.22-μm nylon filter.Finally, the solution was diluted 20 times with 70% aqueous methanol solution before injection into an ultra-performance liquid chromatography coupled to a triple quadrupole mass spectrometer (UPLC-QQQ-MS)(Thermo Fisher Scientific, Waltham, USA).At least two replicates were conducted for each sample.
Separation of free amino acids was performed using a C18 reverse phase column (CORTECS T3 column,120?, particle size: 2.7 μm, length: 100 mm, and internal diameter: 2.1 mm) at 25°C.The binary gradient elution system consisted of 0.1% formic acid-methanol (v/v,phase A) and 0.1% formic acid-water (v/v, phase B).The proportion of A was linearly plotted as follows: 1%(0 min)–1% (1 min)–15% (5 min)–95% (6 min)–95%(7 min)–1% (7.1 min)–1% (9 min).Each sample (2 μL)was injected.The flow rate was adjusted to 0.25 mL min–1.Metabolites were measured using UPLC-QQQ-MS.Mass spectra were recorded using electrospray ionization (ESI).The products, collision energy, and cone voltage were optimized for each amino acid using the SRM scanning model with authentic compounds (Appendix A).
Systematic error was corrected by performing quality control every 2.5 h.
To determine the content of amino acids in transgenic plants ofCsBCATandCsGS, transgenic tobacco leaves were collected.A total of 100 mg of powdered young leaves were weighed and extracted in 0.6 mL of 70%aqueous methanol solution.The metabolites were extracted at 4°C for 12 h, and the supernatant was collected by centrifugation.After passing through a 0.22 μm filter, the extract was submitted to UPLC-QQQMS.The liquid chromatography and mass spectrometry conditions were the same as the method described above, with the exception that 0.1% formic acid-methanol was replaced with pure acetonitrile.
2.3.Association studies
The raw reads of 174 accessions used for genotyping were downloaded from the NCBI database under the project PRJNA595795 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA595795) according to previous research (Zhang W Yet al.2020).Lowquality reads were trimmed with default parameters using Trimmomatic-0.36 (Bolgeret al.2014).The clean RNA-seq reads of 174 accessions were mapped to the DASZ genome.The genotyping pipeline was performed according to a previously described method (Zhang W Yet al.2020).Briefly, 87 298 high-quality SNPs with a minor allele frequency (MAF) greater than 0.05 among these 174 accessions were used to perform genomewide association studies of 17 amino acids in tea plants.Amino acid phenotype data from 174 tea accessions over two years was obtained by performing a targeted metabolomics approach (UPLC-QQQ-MS).A GWAS was conducted using EMMAX Software (Kanget al.2010).Variants were merged into an initial QTL region using the following threshold: i)P-value<0.001; ii) within 500 kb to the lead SNPs; and iii) pairwise LD (r2)>0.1.At least two or more SNPs were present in each merged QTL region.The final QTL region was expanded by 100 kb based on the initial QTL region.The results of a Kyoto Encyclopedia of Genes and Genomes (KEGG)enrichment analysis were visualized in R.Candidate genes were selected based on functional annotation,as well as correlations between phenotype data and gene expression levels.Based on the RNA sequence data of five varieties with the high haplotype and five varieties with the low haplotype, the gene structures ofCsGSandCsBCATwere modified for genotype analysis.Genotyping results ofCsGSandCsBCATwere verified by Sanger sequencing in varieties with high haplotype and varieties with low haplotype (Primers are listed in Appendix B).
2.4.Phylogenetic analysis
To perform the evolutionary analysis of CsGS and CsBCAT, the amino acid sequences of homologous proteins of other species were downloaded from the NCBI Database (http://www.ncbi.nlm.nih.gov/protein).Multiple alignment was performed using the Clustal W tool with default parameters.A phylogenetic tree was built using MEGA7.0 Software with the neighbor-joining method.The numbers on the branches represent the percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1 000 replicates).
2.5.Enzyme activity assays
The open reading frames (ORFs) ofCsGS-L andCsGS-H were cloned into the pMAL-c2X vector fused with a maltose binding protein (MBP) tag at the N-terminal(Appendix B).Recombinant constructs were transformed intoEscherichiacolistrain BL21 (DE3) (Transgen, Beijing,China).The empty pMAL-c2X transformant was used as a control.The positive clone was cultured in ampicillinresistant LB liquid medium at 37°C.Subsequently,isopropyl-β-D-thiogalactopyranoside (IPTG, 1 mmol L–1) was not added to induce the expression of proteins until the OD600value was approximately 0.5.Samples without IPTG induction served as a negative control.The samples were incubated at 16°C for approximately 18 h to induce the expression of MBP-CsGS fusion protein.
Bacterial cells were collected by centrifugation at 4 000 r min–1for 15 min.The sediment was resuspended with 40 mL of CB buffer (20 mmol L–1Tris-HCl (pH=8.0), 0.2 mol L–1NaCl, 1 mmol L–1EDTA).The cell resuspension solution was crushed by a highpressure cell crushing apparatus.To collect the proteins released by the bacteria, the supernatant was collected by centrifugation at 10 000 r min–1for 10 min.The raw protein was purified using dextrin beads 6FF (Smart-Lifesciences, Changzhou, China).Purified proteins were detected by 10% SDS-PAGE analysis.The Bradford Protein Assay Kit (Coolaber, Beijing, China) was used to determine the protein concentration using the Bradford method with bovine serum albumin as a standard(Bradford 1976).The enzyme activity assay was carried out using the method described by Chenget al.(2017)except that the co-enzyme Mg2+was replaced with Mn2+,and the total reaction volume was 1 mL.The content of Gln was measured using UPLC-QQQ-MS.
2.6.Overexpression of CsGS and CsBCAT in model plants
To verify the functions ofCsGSandCsBCAT, the cDNA sequences ofCsGS-L andCsGS-H, as well asCsBCAT-L andCsBCAT-H were isolated from tea plants (Appendix B).Coding sequences of target genes and clonal materials are listed in Appendix C.ORFs ofCsGS-L,CsGS-H,CsBCAT-L andCsBCAT-H were cloned into the pDNOR221 vector.The coding sequence of the target gene was transferred to the plant expression vector pK7WG2D using the gateway cloning system.The resulting constructs were introduced intoAgrobacterium.The positive clone was cultured in LB liquid medium.For stable transformation into wild typeA.thaliana, the strain was collected by centrifugation, and the sediment was re-suspended with the buffer (5% sucrose, 0.5‰Silwet L-77), which made the OD600value 0.8.The floraldipped method was used to transformA.thaliana.After culturing for 16 h in dark, these plants were transferred to normal growth conditions.Three over-expression (OE)lines, identified by qPCR (Appendix B), were prepared to perform metabolite phenotype analysis.
For the transient expression of tobacco leaves, the strains were re-suspended with MMA buffer (10 mmol L–1MES (pH 5.7), 10 mmol L–1MgCl2, 120 μmol L–1acetosyringone) to make the OD6000.8.After incubating for 2 h at 25°C.The solution was injected into the tobacco leaves.Three days later, samples were collected for metabolite extraction and analysis.Seven replicates were performed for each sample.
2.7.Data processing and statistical analysis
All raw files acquired from mass spectrometry were deposited to the Figshare Database (https://figshare.com/s/c53c06d1f4458fabea8b) and processed using TraceFinder 4.0 Software with default parameters, including peak identification, selection, signal integration, and alignment.Briefly, the workflow was as follows.Raw files of samples and authentic standards were imported into TraceFinder 4.0 Software.A data file, containing sample information,retention time, and raw spectra with absolute intensities and peak areas was printed.Metabolites were determined by comparing their mass, retention time, and MS/MS fragmentation patterns against amino acid authentic standards.The mass spectrum of every compound was manually inspected to verify whether the fragment peak was intact.The mean value of each compound was calculated.All raw peak areas were corrected by referring to a method described by Luanet al.(2018) in R.The correlations were analyzed with the Pearson correlation coefficient (PCC) method using the Corrplot the package in R.The correlation between different amino acids was visualized using the pheatmap package in R.Student’st-test and one-way ANOVA were conducted to statistically analyze the data.The significance level for all tests was set atP-value<0.05 (Student’st-test).
3.Results
3.1.Variation in the content of free amino acids in 174 tea accessions
Amino acids make major contributions to the brisk flavor of tea infusion.To investigate the genetic basis of natural variation in amino acids, young leaves of 174 tea accessions were collected in July 2018 (referred to as the YL1 batch) and in April 2019 (referred to as the YL2 batch).Information on these tea accessions is provided in Zhang W Yet al.(2020).We measured the content of free amino acids in the 174 tea plants using UPLC-QQQMS (Appendix D).The content of these amino acids varied.The content of 12 amino acids was lower in 2018 than in 2019, and the opposite pattern was observed for four other types of amino acids (Fig.1).In addition, the relative abundance of Thea was the highest, followed by Asp and Glu, which are closely related to the umami taste of tea infusion.The accumulation of most amino acids was higher in the YL2 batch than in the YL1 batch, which stems from the intense light and high temperature in July that could accelerate carbon metabolism and repress nitrogen metabolism in tea plants.
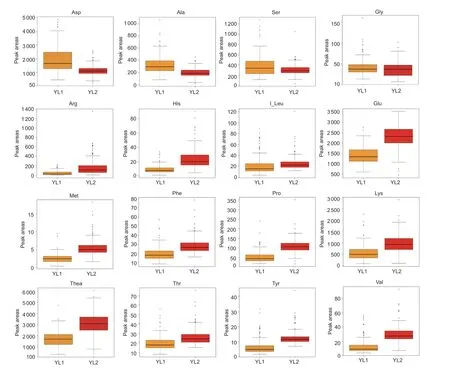
Fig.1 Comparison of the content of amino acids in young leaves (one bud with a leaf, YL) YL1 (collected in July 2018) and YL2(collected in April 2019).The box plot shows the distribution of metabolite.The middle line represents the median.The lower border represents the first quartile.The upper border represents the third quartile.The whiskers of each box indicate the minimum and the maximum.The dots outside the whiskers represent the outliers.I_Leu represents the sum of the Ile peak area and Leu peak area.Asp, aspartic acid; Ala, alanine; Ser, serine; Gly, glycine; Arg, arginine; His, histidine; Ile, isoleucine; Leu, leucine; Glu,glutamate; Met, methionine; Phe, phenylalanine; Pro, proline; Lys, lysine; Thea, theanine; Thr, threonine; Tyr, tyrosine; Val, valine.
Correlations in the content of different amino acids were calculated using PCCs (Appendix E).Most amino acids were highly correlated regardless of whether they were in the YL1 batch or the YL2 batch (Appendix F-a).Strong correlations were observed among some amino acid components.For example, there was a high correlation among I_Leu, tyrosine (Tyr), and Val, and their average correlation coefficients in the YL1 batch and YL2 batch were 0.836 (I_Leu-Tyr), 0.813 (I_Leu-Val),and 0.764 (Tyr-Val), respectively (Appendix F-c).The correlation coefficient between phenylalanine (Phe) and Thr was 0.998 (Appendix F-b).These results indicated that most amino acids might participate in a complex metabolic network, and some amino acids might be involved in the same metabolic pathway.However, weak correlations were observed between the same amino acid in YL1 and YL2 (i.e., Gly1 and Gly2).This result suggests that environmental factors (i.e., light intensity and temperature) might have a greater effect on the accumulation of amino acids in tea plants than in other varieties (Wanget al.2014; Liuet al.2017).
3.2.Genome-wide association studies
Association studies were carried out to identify key genes that regulate variation in the content of amino acids in tea accessions.A total of 1 366 QTLs (–log10(P-value)>3)were identified, and the –log10(P-value) was greater than 5 for 69 of these (Appendix G).A total of 106 QTLs were mapped for the Phe content; 103 QTLs were mapped for the Glu1 and Arg1 traits (Fig.2-A).Potential loci such asalmt12(aluminum-activatedmalatetransporter12) andgs(glutaminesynthetase,W02g005186) were identified(Fig.2-D and E).A total of 138 QTLs were mapped for the I_Leu1 and Val1 traits (Fig.2-A).Several important loci such ascat(cationicaminoacidtransporter8),bact(branched-chainaminoacidaminotransferase),myb108(transcriptionfactorMYB108), andnrt1(nitratetransporter1)were identified (Fig.2-F and G).Overall, QTLs were mostly associated with Lys2, His1 (histidine1), and Phe2;most QTLs are located on Chr1, Chr2, Chr3, Chr6,and Chr7 (Fig.2-A).After the alignment to the DASZ reference genome (Zhang W Yet al.2020), genes located within these QTL regions were registered.A total of 4 191 genes were recorded (Appendix H).Among them,2 619 genes were associated with one amino acid trait;988 genes were associated with two amino acid traits;584 genes were associated with at least three amino acid traits (Fig.2-B).Furthermore, 65 QTLs and 294 genes were identified in both 2018 and 2019viaassociation studies (Fig.2-C; Appendix I).Most of these genes were involved in growth and development-pathways, nitrogen uptake, and amino acid synthesis in higher plants.
3.3.ldentification of genes associated with variation in the content of amino acids
Based on the 4 191 genes identifiedviaassociation studies, KEGG enrichment analysis was carried out(Fig.3).Many genes were enriched in the following pathways: “Metabolic pathway,” “Biosynthesis of secondary metabolism,” “Carbon metabolism,” “Arginine and proline metabolism,” “Tryptophan metabolism,” and“Phenylalanine metabolism.” Thirty potential genes were retained.Detailed information including gene ID,annotation, associated metabolic traits, position, –log10(Pvalue), and correlation coefficients are provided in Table 1.
Glutamate metabolism is a key biological process in plants.Here, we identified several genes associated with the glutamate contentviaassociation studies (Table 1;Appendix H).At thegslocus on chromosome 2, we identified a gene that encodes a glutamine synthetase(W02g005186).It was associated with levels of Glu1(P=3.71×10–4) and Arg1 (P=4.61×10–5).In addition, thegadlocus on chromosome 4 (W04g010842,P=2.46×10–5)was associated with the Asp content.This gene encodes a glutamate decarboxylase, which was involved in the conversion of Glu to 4-aminobutanoate.At thee2fblocus,W15g032954encodes an E2FB transcription factor,which was associated with several amino acid traits,namely Ala2 (P=2.69×10–6), Ser2 (P=1.30×10–5), Asp2(P=6.01×10–8), and Glu2 (P=6.7×10–7).
Humans and animals cannot synthesize BCAAs by themselves, and obtain them exclusively from plants.Therefore, plants are the main source of BCAAs for humans and animals.In this study, we identified several genes that might be involved in the metabolic pathways associated with BCAAs (Table 1; Appendix H).Thebactlocus (W07g016468) was located on Chr7, and it was associated with Val1 (P=4.67×10–5) and I_Leu1(P=3.56×10–6).It encodes a branched-chain amino acid aminotransferase and plays a role in the generation of Val, Ile, and Leu using 3/4-methyl-2-oxobutyrate as a substrate (Binder 2010).TheW14g030305gene encodes an isochorismate synthase.TheW02g004515gene encodes an elongation factor 1-delta protein that might be involved in plant growth and development.Both of them were associated with Val1 (P=2.86×10–7,P=3.79×10–4)and I_Leu1 (P=6.23×10–6,P=9.55×10–5).W11g023081,W11g023082, andW11g023084encode bZIP transcription factors.They were identified to be associated with Val1(P=1.32×10–4) and I_Leu1 (P=1.64×10–4).At thecatlocus, the geneW05g011891encodes a cationic amino acid transporter.It was also associated with four amino acid traits (Tyr1,P=1.02×10–5; Val1,P=8.81×10–4; His1,P=8.28×10–4; I_Leu1,P=2.45×10–4).
Nitrate transporters (NRTs) and ammonia transporters are important mediators of nitrogen uptake and nitrogen utilization in plants.Amino acid permeases (AAPs) are important carrier proteins in amino acid metabolism in plants.In our study, we identified 24 NRTs, three amino acid permeases and two ammonia transportersviaassociation studies (Table 1; Appendix H).
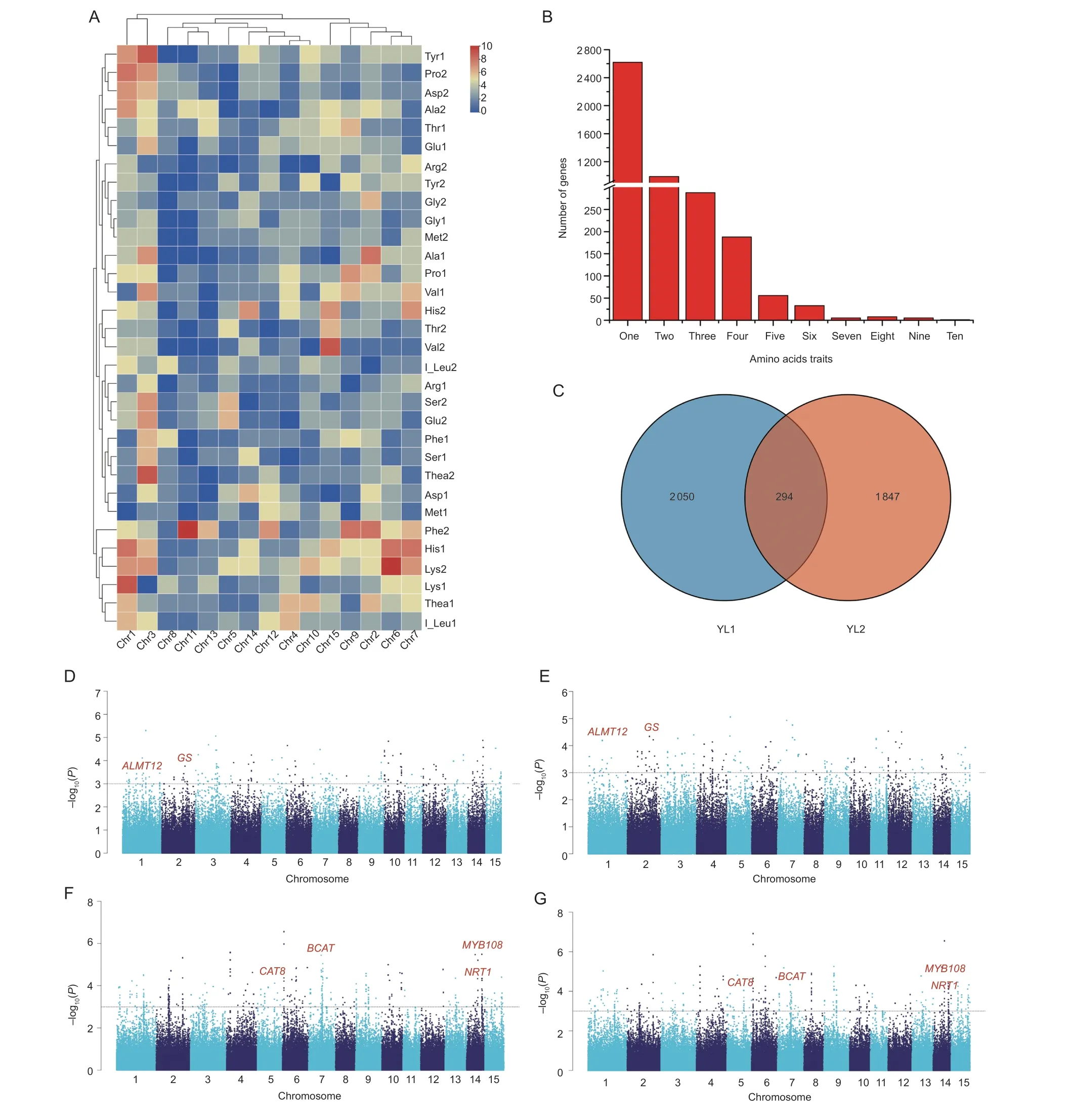
Fig.2 Statistics of genome-wide association study (GWAS) results.A, chromosomal distribution of QTLs associated with amino acid traits.‘1’ represents the YL1 batch, and ‘2’ represents the YL2 batch.B, histogram distribution of genes associated with amino acid traits.C, Venn diagram of the common genes shared in YL1 and YL2 via GWAS.YL1 and YL2, young leaves (one bud with a leaf, YL) collected in July 2018 and April 2019, respectively.D–G, Manhattan plots of Glu1 (D), Arg1 (E), I_leu1 (F), and Val1 (G)traits in tea plants under the MLM model.ALMT12, aluminum-activated malate transporter 12; GS, glutamine synthetase; BCAT,branched-chain amino acid aminotransferase; CAT8, cationic amino acid transporter 8; MYB108, transcription factor MYB108; NRT1,nitrate transporter 1.The gray dashed horizontal line represents the significance threshold (P-value<1×10–3, –log10(P-value)=3).
3.4.Genotype analysis of CsGS and CsBCAT
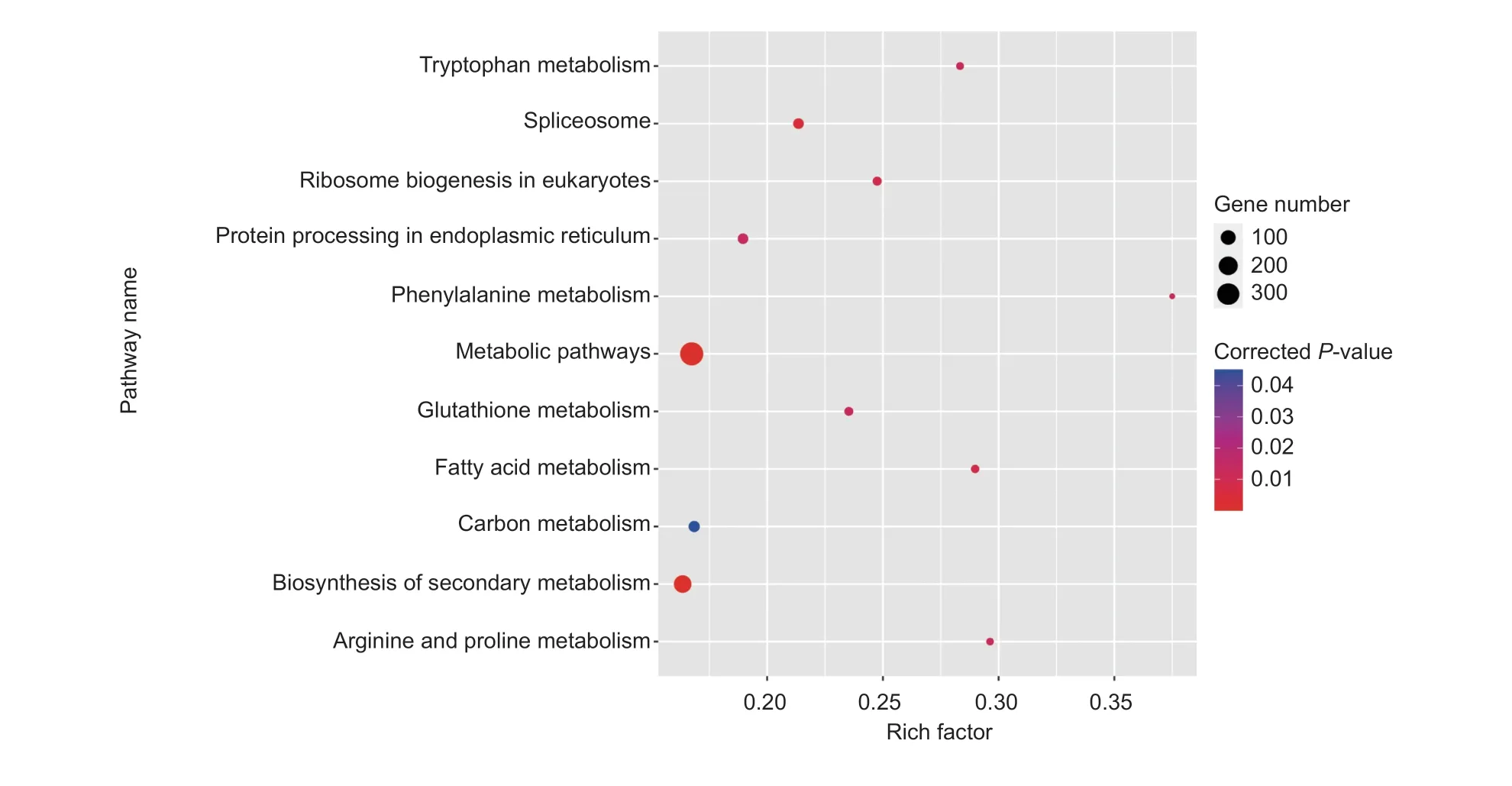
Fig.3 KEGG enrichment analysis of the genes identified by a genome-wide association study (GWAS) (P-value<0.05).
Two candidate genes,W02g005186(CsGS; Glu1,P=3.71×10–4; Arg1,P=4.61×10–5) andW07g016468(CsBCAT; Val1,P=4.67×10–5; I_Leu1,P=3.56×10–6),were used to conduct genotype analysis; these genes are involved in the Gln biosynthesis pathway and BCAA pathway, respectively (Fig.4-A and C).First, the structures ofCsGSandCsBCATwere re-annotated by aligning RNA sequencing reads to the DASZ genome (Zhang W Yet al.2020).Genotype analysis results showed that one non-synonymous SNP (c.A1054C, “c” represents coding sequence; p.I352L, “p” represents protein sequence) was identified in the coding sequence ofCsGS(Fig.4-B),and three genotypes were identified.T/G conferred the highest level of Glu1 and lower levels of Arg1 (N/N indicates heterozygous SNPs), whereas G conferred the lowest level of Glu1 and higher levels of Arg1, and T represents the intermediate phenotype (Fig.4-E).Three non-synonymous substitutions in the coding sequence ofCsBCAT(c.G512A, p.R171Q; c.G767A, p.R256H;c.T1055C, p.V352A) were used to perform genotype analysis in 174 tea accessions.Six different genotypes were generated (Fig.4-D).The CCA genotype conferred the highest levels of Val1 and I_Leu1, and the genotype C/TC/TA conferred the lowest levels of Val1 and I_Leu1(Fig.4-F).Furthermore, Sanger sequencing was performed in varieties with high haplotype and several cultivars with low haplotype to verify the genotype at SNP1054ofCsGSand the genotypes at SNP512, SNP767, and SNP1055ofCsBCAT.The Sanger sequencing results ofCsGS(Appendix J-a)andCsBCAT(Appendix J-b) were consistent with the genotyping results ofCsGSandCsBCATin tea accessions.Therefore, we selected the genotypes G (namedCsGS-L, G)and T/G (namedCsGS-H, T) ofCsGSand the genotypes CCA (namedCsBCAT-H, CCA) and C/TC/TA (namedCsBCAT-L, TCA) ofCsBCATto perform functional analysis.
Phylogenetic analysis showed that CsGS-L and CsGS-H were clustered with CsGS1.1, CsGS1.2 and CsGS1.3 (Appendix K-a), while CsBCAT-L and CsBCAT-H were closely associated with AtBCAT4.1 and AtBCAT5.2(Appendix K-b).
3.5.Overexpression of CsGS and CsBCAT alleles alters the accumulation of free amino acids
To verify the molecular functions ofCsGSandCsBCAT,transient transformation ofN.benthamianaleaves was performed.The content of free amino acids in transgenic tobacco leaves was measured using UPLC-QQQ-MS(Appendix L).PCA revealed that the metabolite profiles induced by the two allelesCsGSandCsBCATwere distinct (Fig.5-A and E).This result indicates that the two alleles ofCsGSorCsBCATplayed different roles in the accumulation of free amino acids.The Glu content in transgenic tobacco leaves was significantly lower inCsGS-L than that inCsGS-H (Fig.5-B).Conversely,the content of Gln and Arg in transgenic tobacco leaves was significantly higher inCsGS-L than that inCsGS-H(Fig.5-C and D).This result is consistent with the result of genotype analysis ofCsGSin the 174 tea accessions.Thirteen amino acids were accumulated differentially in the transgenic tobacco leaves ofCsGS-L andCsGS-H, with the exception of Glu and Gln (Appendix M).The transient overexpression ofCsBCAT-L andCsBCAT-H in tobacco leaves reduced Thr content and induced the accumulation of Ile, Leu, and Val (Fig.5-F, G, H, and J).These results indicate thatCsGSandCsBCATmight play a key role in regulating the accumulation of free amino acids.
InA.thaliana, the overexpression ofCsBCATresultedin differential accumulation of 17 amino acids (Appendices N-a and O).As shown in Appendix N-b, most of these amino acids accumulate to a greater degree in the transgenic lines ofCsBCAT-H compared withCsBCAT-L.The content of Ile, Leu, and Val was significantly higher inCsBCAT-H lines than inCsBCAT-L transgenic lines (Appendix N-c–e).
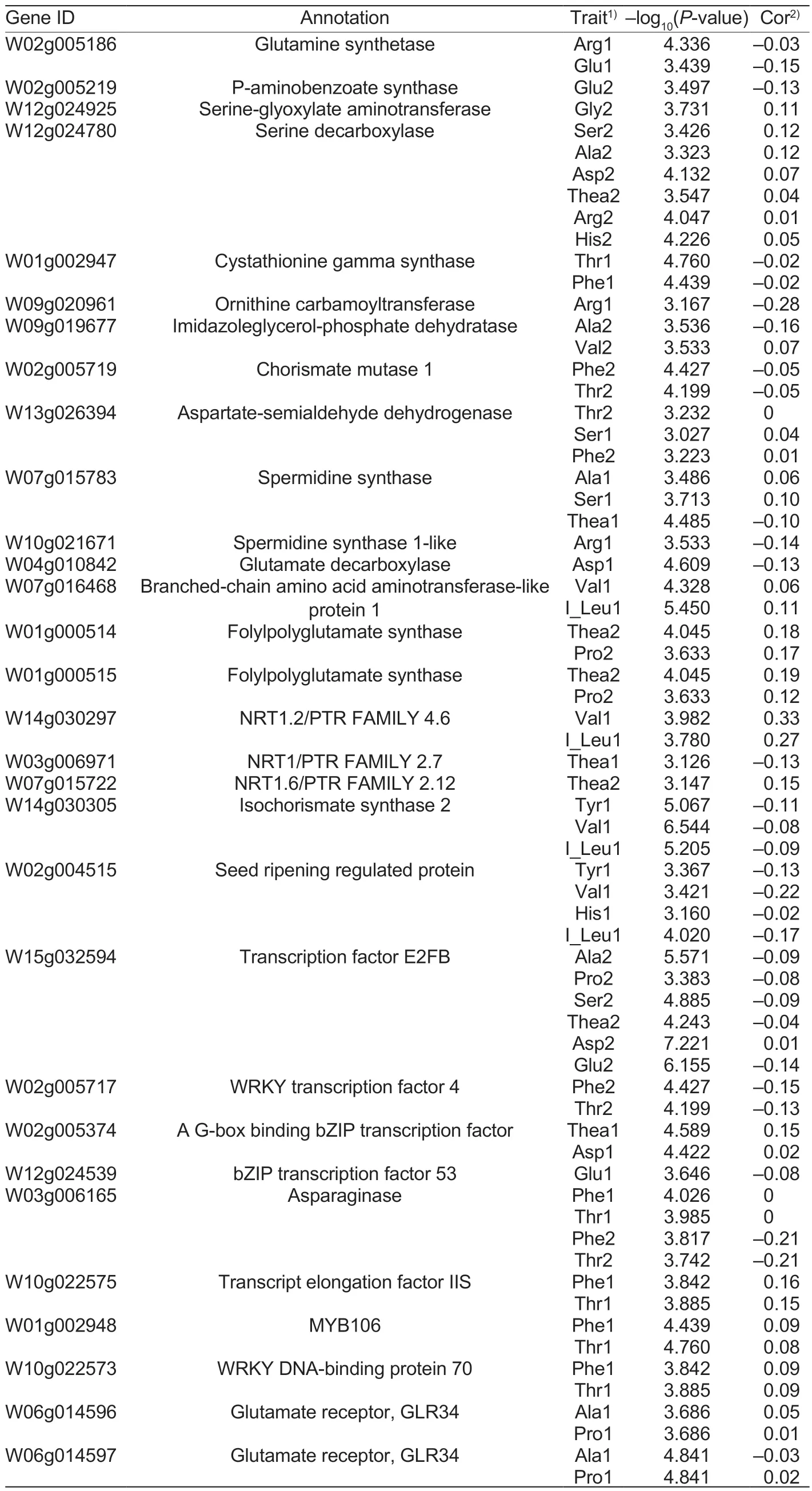
Table 1 Potential genes associated with the content of amino acids
3.6.Enzyme activity analysis of CsGS alleles involved in the synthesis of Gln
To assess the effects of nonsynonymous substitutions on the catalytic activity of CsGS-L and CsGS-H,enzyme activity assays were performed.The coding sequences ofCsGS-L andCsGS-H were cloned into the prokaryotic expression vector pMAL-c2X and fused to an MBP tag at the N terminal.The recombinant constructs were transformed intoE.colistrain BL21 (DE3)host cells, and the empty vector pMAL-c2X was used as a negative control.The purified protein was resolved by SDS-PAGE analysis.The molecular weight of MBPCsGS-L and MBP-CsGS-H was approximately 84 kDa,which is consistent with the calculated molecular weight(Fig.6-A).Both CsGS-L a n d C s G S-H p r o t e i n s could catalyze Glu and free ammonium into Glninvitro(Fig.6-B; Appendix P).Nevertheless, the level of Gln catalyzed by CsGS-L was significantly higher than that catalyzed by CsGS-H(F i g.6-C).T h i s r e s u l t suggests that a nonsynonymous substitution in SNP1054of CsGS could affect the catalytic activity of CsGS-L and CsGS-H and thus Gln generation.
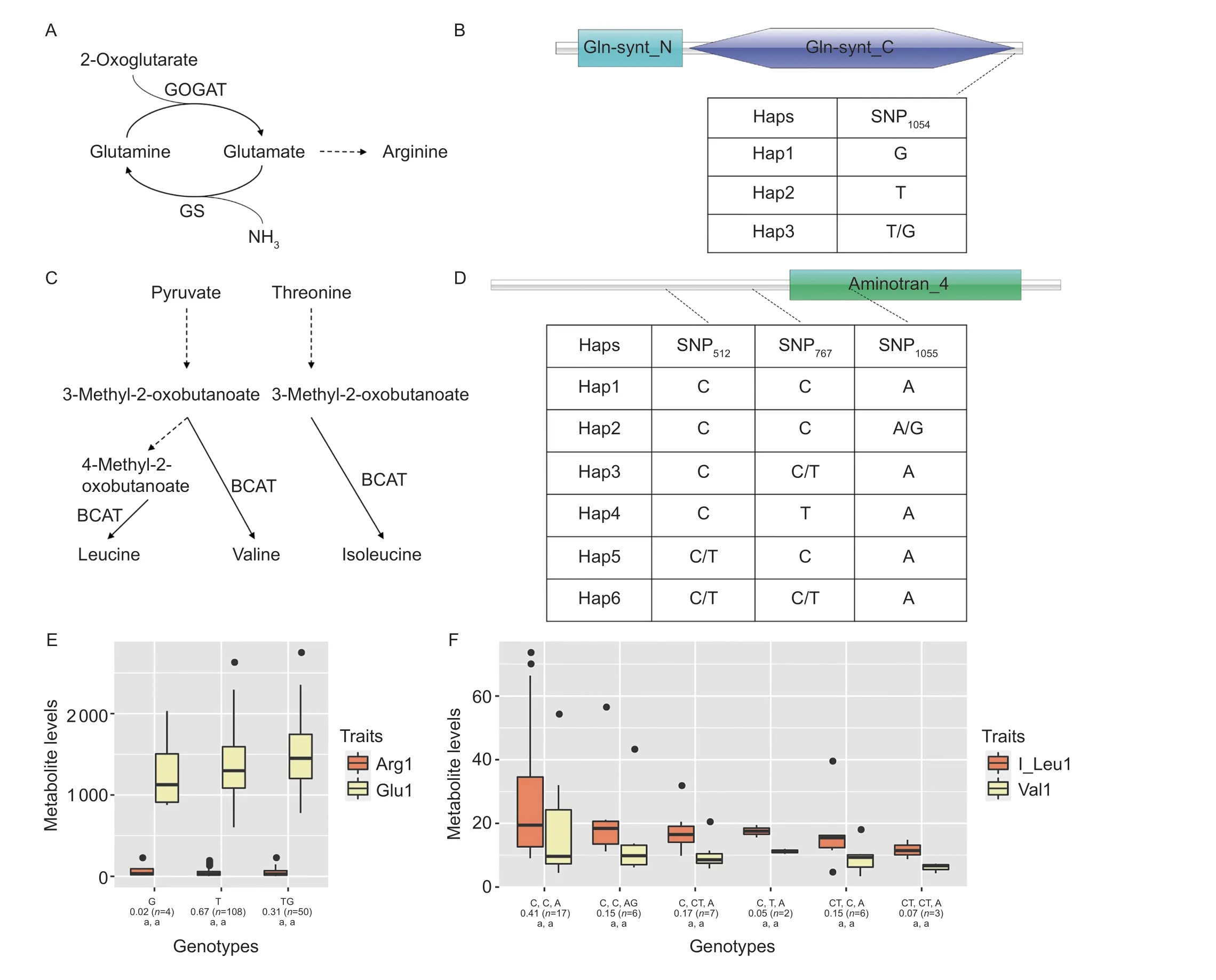
Fig.4 Genotype analysis of CsGS and CsBCAT.A, biosynthesis pathway of Glu.B, haplotypes of CsGS (Hap1–Hap3) in 174 tea accessions.The indigo rectangle and the purple hexagon indicate the conserved elements.C, biosynthesis pathway of BCAA.D,haplotypes of CsBCAT (Hap1–Hap6) in 174 tea accessions.The green rectangle indicates the conserved element.E, box plots of the genotype analysis using non-synonymous SNPs in CsGS.F, box plots of the genotype analysis using non-synonymous SNPs in CsBCAT (each SNP site is separated by a comma).In E and F, ‘n’ represents the sample size of this genotype in the tea population.The letters attached on the bottom of the x-axis show the levels of significance according to Tukey’s test (adjusted P<0.05; two-sided).
4.Discussion
4.1.GWASs helped identify loci involved in regulating variation in the content of amino acids in tea plants
The tea plant is an economically important crop, and the young leaves are the most economically significant parts of tea plants.Amino acids are nitrogenous compounds that are enriched in the young leaves of tea plants and contribute greatly to the umami and sweet taste of tea infusions (Yu and Yang 2020).For example, theanine contributes to the formation of 2,5-dimethyl pyrazine, which confers a key roasted peanutty flavor in oolong tea (Yu and Yang 2020).The content of amino acids in the fresh leaves of tea plants is also related to the suitability of tea varieties for production.For example, cultivars with high levels of free amino acids are suited for green tea production,whereas cultivars with a high content of polyphenol are suited for the production of black tea and Pu-erh tea (Meiet al.2021).Therefore, studying the natural variation in the content of free amino acids in tea plants is important for characterizing the adaptability of different tea varieties.
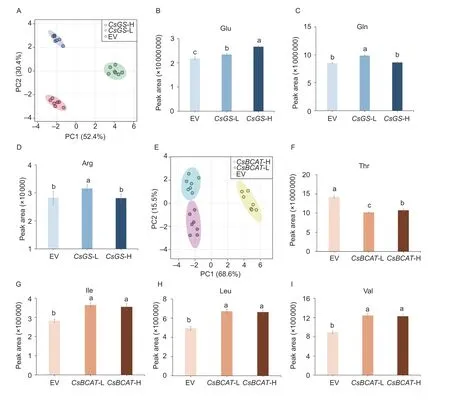
Fig.5 Accumulation profile of amino acids in CsGS and CsBCAT transgenic tobacco leaves.A, PCA of the profile of accumulated metabolites in transgenic tobacco leaves of empty (EV), CsGS-L and CsGS-H.B–D, comparison of the content of Glu, Gln, and Arg in transgenic tobacco leaves of CsGS-L and CsGS-H with the EV as a negative control.E, PCA of the profile of accumulated metabolites in transgenic tobacco leaves of EV, CsBCAT-L and CsBCAT-H.F–I, analysis of Thr, Ile, Leu, and Val levels in transgenic tobacco leaves of EV, CsBCAT-L and CsBCAT-H.The data are expressed as the mean±SE (n=7); lowercase letters indicate P-values less than 0.05 according to a t-test.
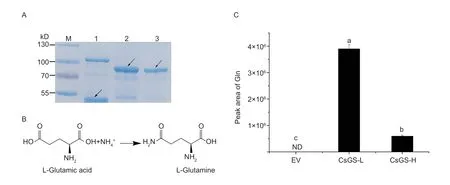
Fig.6 Enzyme activity assays of CsGS-L and CsGS-H for the generation of Gln.A, SDS-PAGE analysis of the purified proteins.M: protein marker; lane 1, the empty vector, pMAL-c2X; lane 2, MBP-CsGS-L; lane 3, MBP-CsGS-H.The arrow points to the target protein band.B, the biosynthesis pathway of Glu to Gln.C, identification of the Gln level generated from CsGS-L and CsGS-H using Glu as a substrate in vitro.ND indicates not detected.Data are mean±SE (n=4); lowercase letters (a, b, c) indicate P-value less than 0.05 according to a t-test.
The content of amino acids is a quantitative trait.Forward genetics is an effective approach for studying quantitative traits in higher plants.Unlike model plants or other plants with genetic transformation systems,C.sinensisis a self-incompatible perennial woody plant,which means that it is not practical for the study of the quantitative traits of tea plants by constructing BC1, RIL,NILs, and SSSLs.Therefore, an F1generation population was constructed, and QTL mapping was performed to study metabolite traits in tea plants, such as amino acids, catechins, caffeine and theobromine.Quantitative traits are generally controlled by micro-effect polygenes in plants.However, factors such as population size,recombination events, and genetic heterogeneity can affect the ability of QTL mapping to identify loci or genes associated with metabolite traits in tea plants (Price 2006).GWASs were performed using high-density SNPs,which makes up for the deficiency of QTL mapping.In this study, 87 298 high-quality SNPs were recorded, which is far more than the number of genetic markers used in QTL mapping (Maet al.2018; Xuet al.2018; Huanget al.2022).Multi-year or -position repeats tend to identify loci associated with target traits and the environment (Price 2006).Wang performed a GWAS and identified 27 stable SNPs associated with the timing of spring bud flush (TBF)over three years (Wanget al.2019).We measured amino acid content in 174 tea plants over two years.However,the accumulation of free amino acids differed between YL1 and YL2.The strength of the correlations between amino acids in YL1 and YL2 was lower than that reported in a previous study (Huanget al.2022), which might be explained by the fact that their samples were collected in the same season (spring) in 2018 and 2019, whereas our samples were collected in different seasons.
To date, some genes involved in the metabolic pathways of theanine, ethylamine, and glutamate have been isolated, yet many genes remain unknown.After mapping the transcriptome data sets of 174 tea accessions to the genome of DASZ, 87 298 high-quality SNPs were obtained, and they were used to genotype the 174 tea accessions.The amount of data in this study was much larger than that in Fanget al.(2021),which was aimed at identifying functional genetic variants using a high-density genetic map.Based on the GWAS results, nitrate transporter 1/peptide transporter (NRT1/PTR) proteins, such as those encoded byW14g030297,W03g006971, andW07g015722, were identified to mediate the uptake and transfer of nitrate from soil to the entire plant (Wanget al.2014).Ammonium transporters(W07g016454,W13g027892) are responsible for NH4+assimilation (Haoet al.2020).Tetrahydrofolate (THF)is an essential co-factor that acts as a carbon (C1)carrier catalyzing the formation of amino acids, such as Met, Gly, and Ser (Hanson and Gregory 2011).Here,three folylpolyglutamate synthases (W06g013998,W01g000514, andW01g000515) in tea plants were identified, and these might regulate the generation of tetrahydrofolate-Glu (n) by linking Glu residues to folate(vitamin B9) (Srivastavaet al.2011).The addition of poly-glutamate to folate could improve its co-enzyme affinity and stability (Hanson and Gregory 2011).In higher plants, amino acids are loaded and transported between different tissues or organs through the xylem and phloem.This redistribution is inseparable from the activity of amino acid transporters.Our study also identified genes encoding amino acid transporters(Fischeret al.1998) such as amino acid permeases(AAPs), auxin transporters (AUXs), neutral amino-acid transporters (ANTs), and cationic amino-acid transporters(CATs) (Table 1; Appendix H).Phenylalanine is an essential primary metabolite that connects the primary metabolic pathways and secondary metabolic pathways.Phenylalanine, the downstream product of the shikimate pathway, plays a role in the biosynthesis of flavonoids,pro-anthocyanidins and catechins (Maeda and Dudareva 2012), which are important secondary metabolites in tea.Here, six phenylalanine ammonia lyases were identifiedviaa GWAS (Appendix H), and these were verified by the catalysis of Phe to cinnamate (Wuet al.2017).
4.2.Mechanism underlying variation in glutamate and branched-chain amino acids in tea plants
Higher plants utilize two isozymes of glutamine synthetase(cytosolic GS1 and chloroplastic GS2) to assimilate nitrogen.GS1 is mainly responsible for nitrogen assimilation in the plant roots, whereas GS2 functions in the re-assimilation of ammonium in chloroplasts and ammonium released from photorespiration (Lamet al.1995; Tairaet al.2004).CsGS1.1, CsGS1.2, and CsGS1.3 are cytosolic GS1 proteins, suggesting that CsGS in this study might be involved in the nitrogen assimilation process in the roots of tea plants (Fuet al.2021).It has been demonstrated thatGS1inO.sativanot only plays a role in enhancing the accumulation of free amino acids, but also regulates the total nitrogen content (Caiet al.2009).In addition, GS1 regulates plant growth and ammonium homeostasis (Moisonet al.2011), as well as reassimilation of ammonium released during protein catabolism (Moreiraet al.2022).Here, the overexpression ofCsGSin tobacco leaves promotes the accumulation of Gln and many other amino acid components.These results suggest thatCsGS(W02g005186) might facilitate the accumulation of free amino acids by enhancing ammonium re-assimilation in tea plants.Non-synonymous substitutions of SNP1054inCsGSalter the accumulation of free amino acids in transgenic plants.This indicates that SNP1054might be important for the catalytic activity of CsGS in the synthesis of Gln, and it could be important locus for clarifying the nitrogen utilization of tea plants with different genotypes.
BCAAs are synthesized only in plants; they thus need to be consumed by mammals to meet their nutritional requirements (Dulliuset al.2020).In plants,BCAT is responsible for regulating the accumulation of BCAAs.BCAT regulates the catabolism of BCAAs in the mitochondria (Binder 2010; Maloneyet al.2010;Leeet al.2019), and BCAT stimulates the biosynthesis of BCAAs in the chloroplast (Binder 2010; Maloneyet al.2010).In our study,CsBCAT(W07g016468) was identified to be associated with the content of Val1 and I_Leu1viaa GWAS.Overexpression ofCsBCATin tobacco leaves andArabidopsissignificantly increased BCAA content.Moreover,CsBCAT-L andCsBCAT-H differentially regulated the accumulation of free amino acids in transgenic plants.This indicates thatCsBCATisolated in our study might be responsible for BCAA biosynthesis in tea plants, and SNPs (c.G512A, p.R171Q;c.G767A, p.R256H; c.T1055C, p.V352A) might play a key role in its catalytic activity.InO.sativa, the expression ofBCATwas affected by aluminum (Jinet al.2019) and nitrogen conditions (Sunet al.2020) in soil.Tea plant is a nitrogen-preferring and aluminum-richness plant.This suggests thatCsBCATmight regulate the growth and development of tea plants in response to nitrogen conditions and the content of aluminum in soil.
Overall, these findings provide new insights that will aid studies of the genetic mechanism underlying variation in the content of free amino acids in tea plants, molecularassisted breeding, and the identification of elite genes for improving the quality of tea in the future.
5.Conclusion
We determined the content of free amino acids in young leaves from a collection of tea germplasms using an UPLC-QQQ-MS system.After alignment of transcriptome data from 174 tea accessions to the DASZ genome,87 298 high-quality SNPs (MAF>0.05, missing rate<0.2)were identified.GWASs were conducted based on the amino acid phenotype data and genotype data of 174 tea accessions.A total of 1 366 loci were identified, and 69 loci (–log10(P-value)>5) were analyzed.KEGG enrichment analysis revealed that the genes screened from these loci were mostly enriched in the following pathways: “metabolic pathway,” “biosynthesis of secondary metabolism,” “carbon metabolism,” “arginine and proline metabolism,” “tryptophan metabolism,” and “phenylalanine metabolism.” Finally, the functions of two loci,gs(W02g005186,CsGS) andbcat(W07g016468,CsBCAT), were verified.The high-genotype and low-genotype ofCsGSdifferentially regulated the accumulation of free amino acids in transgenic tobacco leaves, and their proteins differentially catalyzed the formation of Gln.There was no significant phenotypic difference between the high-genotype and low-genotype ofCsBCATin transgenic plants.However, the overexpression ofCsBCATinArabidopsisand tobacco leaves promoted the accumulation of Val, Ile, and Leu.In addition, the content of amino acids in tea plants might be affected by external factors; phenotype data need to be collected for several consecutive years to confirm this possibility.In conclusion,GWASs can aid the molecular breeding of tea plants and facilitate the identification of elite genes to enhance the abundances of key compounds associated with tea flavor and the health benefits of tea plants.
Acknowledgements
We thank Dr.Sun Zheng and Dr.Huang Zhiheng (National Key Laboratory for Germplasm Innovation & Utilization of Horticultural Crops, Huazhong Agricultural University,China) for offering assistance on protein purification.This work was supported by the Huazhong Agricultural University Scientific & Technological Self-Innovation Foundation, China (2017RC002).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.10.002
 Journal of Integrative Agriculture2023年11期
Journal of Integrative Agriculture2023年11期
- Journal of Integrative Agriculture的其它文章
- Germplasm and molecular breeding in horticultural crops
- Development and application of KASP marker for high throughput detection of the seedless trait in grapevine
- QTL analysis of early flowering of female flowers in zucchini(Cucurbita pepo L.)
- A novel mutation in ACS11 leads to androecy in cucumber
- Comprehensive analysis of the full-length transcripts and alternative splicing involved in clubroot resistance in Chinese cabbage
- Virucidal activity of MICRO-CHEM PLUS against African swine fever virus