
Analysis of the chloroplast genomes of four Pinus species in Northeast China: Insights into hybrid speciation and identif ication of DNA molecular markers
2022-11-04 09:23:26TaoYuZhiyuanJiaBuddhiDayanandaJunqingLiXiaoleiGuoLiangShiXiaowenYuanYanGao
Journal of Forestry Research 2022年6期
Tao Yu · Zhiyuan Jia · Buddhi Dayananda ·Junqing Li · Xiaolei Guo · Liang Shi ·Xiaowen Yuan · Yan Gao
Abstract Species of the Pinus genus provide a classical model for studying hybrid speciation. Although studies on two narrowly distributed species ( Pinus funebris and P. takahasii) concluded that they originated from two widespread species ( P. sylvestris and P. densif lora) via hybrid speciation,the conclusion was based on a low number of informative restriction sites. In this study, we analyzed the sequences of four Pinus chloroplast (cp) genomes ( P. sylvestris, P. densif lora, P. funebris and P. takahasii) to clarify whether hybrid speciation was involved. The complete cp-genomes of Pinus species ranged in size from 119,865 to 119,890 bp, similar to other Pinus species. Phylogenetic results based on the whole cp-genomes showed P. sylvestris clustered with P.funebris and P. takahasii, which suggested that P. sylvestris was the paternal parent in hybridization events. In an analysis of simple sequence repeats (SSRs), we detected a total of 69 SSRs repeats among the four Pinus cp-genomes;most were A or T bases. In addition, we identif ied divergent hotspot regions among the four Pinus cp-genomes ( trnE
Keywords Pinus · Chloroplast genomes · Hybrid speciation · Divergence hotspot regions · Phylogenetic relationship
Introduction
Hybridization plays a signif icant role in plant evolutionary processes (Yakimowski and Rieseberg 2014 b). Polyploidy and homoploid hybrid speciation off er a rapid strategy for plants to adapt to diff erent environmental conditions (Gross and Rieseberg 2005). Polyploidy is involved in the evolutionary dynamics of adaptation and speciation in many higher plants (Hegarty and Hiscock 2008). In maize, olive tree and bread wheat, natural hybridization events and speciation that resulted from allopolyploidization have been reported (Gault et al. 2018; Julca et al. 2018; Seixas et al.2018; Wicker et al. 2018). Unlike the off spring of polyploid hybrids, the off spring of homoploid hybrids retain their parental chromosome number, which remains more porous and less likely to undergo speciation (Vallejo - Marín and Hiscock 2016). The most iconic examples of homoploid hybrids are fromHelianthusandPinaceaespecies (Watano et al. 2004; Staton et al. 2009; Yakimowski and Rieseberg 2014 a). However, compared withHelianthus,Pinaceaehomoploid hybrids research are rare.
Pinaceaeis the largest group of gymnosperms consisting of about 230 species in 10 genera, mainly distributed in the northern hemisphere (Kim et al. 2017). SeveralPinusspecies are hypothesized to have a hybrid origin, but the history of origin has only been rigorously proven for a few species,such asP. tabulaeformisandP. yunnanensisin Southwest China (Gross and Rieseberg 2005).Pinaceaespecies are abundant in Northeast China, and a phylogenetic study ofP. funebrisandP. takahasiiin that area revealed a possible origin via homoploid hybrid speciation betweenP. sylvestrisandP. densif lora(Ren et al. 2012). However, a lack of eff ective molecular markers has hindered the elucidation of the speciation mechanism of these species.
To understand the molecular phylogeography and population genetic structure ofPinusspecies eff ectively, genetic markers are necessary (Savolainen et al. 2013). However,due to the nature of its large genome size, it is diffi cult to develop ample nuclear markers (Zonneveld 2012). Instead,chloroplast (cp) genomes are small and easier to obtain homologous genes to provide comparable genetic information (Daniell et al. 2016; Yu et al. 2020). With the development of next-generation sequencing (NGS) techniques,obtaining the cp-genome information has become more convenient. Typically, the cp-genome of seed plants comprises 120-210 kb, has a highly conserved genomic structure, multiple copies, and low molecular mass (Curci et al.2015; Ruhsam et al. 2015). The cp-genomes in most plants are uniparentally inherited maternally, whereas they are uniparentally paternally inherited inPinusspecies, which could provide a diff erent perspective to study hybrid speciation(Zeb et al. 2019).
In this study, we sequenced the entire cp-genome of fourPinusspecies (P. sylvestris,P. densif lora,P. funebrisandP. takahasii) to identify genes and comparatively analyze their cp-genomes.P. densif lorais widely distributed in East Asia, including Northeast China, Korea, and Japan, and is the most important and popular coniferous species in the Shandong Peninsula, China and Korea, whileP. sylvestrisis widely distributed in Europe and eastern Asia (Ren et al.2012; Kang et al. 2019). In contrast to the more widely distributed and plantedP. sylvestrisandP. densif lora,P. funebrisis endemic and restricted to an area between 800 and 1600 m in the eastern Changbai Mountains in Jilin Province,andP. takahasiiis restricted to Xingkai Lake area in Heilongjiang Province (Xue et al. 1990). In this study, we aimed to (1) analyze the cp-genome structural characteristics of thePinusspecies, (2) identify simple sequence repeat (SSR)loci to provide resources to study the population genetic structure and phylogeography ofPinusspecies, (3) identify divergence hotspots in cp-genomes as potential DNA barcodes; and (4) infer the phylogenetic relationships among thePinusspecies using the complete cp genomes and discuss homoploid hybrid speciation.
Materials and methods
Sampling, DNA extraction, and sequencing
Fresh leaves ofP. sylvestris(Honghuaerji Nature Reserve,48°15′55″ N, 119°59′05″ E, 756 m a.s.l.),P. densifl ora(Kunyushan Nature Reserve, 37°16′31″ N, E 121°46′3″ E,191 m a.s.l.),P. funebris(Changbaishan Nature Reserve,42°24′21.06″ N, 128°6′31.85″ E, 717 m a.s.l.), andP. takahasii(Xingkai Lake National Nature Reserve, 45°16′2.23″N, 132°42′20.47″ E, 72 m a.s.l.) were collected, and total genomic DNA was extracted using the CTAB method(Doyle 1987). Libraries were prepared in accordance with the manufacturer’s instructions with a 350-bp average insert size and a paired-end f low cell using Illumina HiSeq 2500 platform (2 × 150-bp read length) (Biomarker Co, Beijing,China).
Chloroplast genome assembly and annotation
Raw data were trimmed with a f ilter standard (Q ≤ 5 or N base content > 10%). Afterward, cp-sequence reads were extracted by the Burrows-Wheeler Aligner (BWA)(Peters et al. 2011) using the cp-genome ofP. densif lora(MK285358) (Kang et al. 2019) as the reference. We used NOVOPlasty 4.0 (Dierckxsens et al. 2016) withk-mer length 31 and thepsbAgene ofP. densif loraas a seed for the assembled circular genome for four species, and all other parameters were set as the default options. The entire sequences of the four cp-genomes were annotated using CpGAVAS (Liu et al. 2012) and the default options. Each gene was then verif ied manually by comparison with those of the cp-genomes of other pine species through a BLASTN search (Chen et al.2015) to choose highly similar sequences in the NCBI database ( https:// www. ncbi. nlm. nih. gov/ nucco re/? term= compl ete% 20chl oropl ast% 20gen ome ) using BLAST. A circular cpgenome map was drawn using Organellar Genome DRAW(ORDRAW) version 1.3.1 (Lohse et al. 2007).
Simple sequence repeat (SSR) analysis and codon usage of the chloroplast genomes
SSRs were predicted using the MISA perl script (Institute of Plant Genetics and Crop Plant Research, Gatersleben,Germany) to identify perfect SSRs inP. funebris,P. takahasii,P. sylvestris, andP. densif lora.The search parameters were set to identify a minimum of 10 repeats for mononucleotide SSRs, eight repeats for dinucleotide, four repeats for trinucleotide, four repeats for tetranucleotide, three repeats for pentanucleotide, and three repeats for hexanucleotide SSRs (Asaf et al. 2018).Relative synonymous codon usage (RSCU) was determined using MEGA 7.0.21 (Kumar et al. 2016) and a cluster analysis using the maximum distance method, shortest distance method, and average method in R version 4.0.2 (R Team 2013).
Sequence divergence analysis in four Pinus chloroplast genomes
The complete cp-genomes for the fourPinusspecies determined here (P. densifloraMT786135,P. sylvestrisMT796488,P. takahasiiMT786134,P. funebrisMT793600), two forP. sylvestris(MT796488, KR476379.1),and four forP. densiflora(JN854210.1, MK285358.1,MF990371, MT786135) were aligned using MAFFT version 7 with default parameters (Katoh et al. 2005). The indel polymorphisms were identif ied using a sliding window analysis and DnaSP 5 (Librado and Rozas 2009) with a step size of 200 bp and window length of 600 bp. To better reveal the structural features ofP. sylvestris rps8andpsbBgenes, we used the online tool Multiple Expectation Maximization for Motif Elicitation (MEME Suite 5.1.1; Bailey et al. 2009)to predict conserved motifs. The positive selection nonsynonymous to synonymous ratio (Ka/Ks) ofrps8andpsbBwas calculated using DnaSP 5 (Librado and Rozas 2009).
Phylogenetic analysis
For the phylogenetic analysis of the cp-genomic sequences of 25Pinusspecies and three other gymnosperm species(Abies koreana,Abies sibirica,andTaxus baccata) as outgroups, the cp-genomes were aligned using MAFFT (Katoh et al. 2005). Phylogenetic analyses were performed using maximum likelihood (ML) and maximum parsimony (MP)methods on MEGA 7.01 (Kumar et al. 2016) with 1000 bootstrap replicates. For ML analyses, the best-f it model is general time reversible, with gamma distribution (GTR + G)according to the Akaike information criterion (AIC) suggested by Modeltest in MEGA7.
Results and discussion
Features of four Pinus species chloroplast genomes
Complete plastid genomes of fourPinusspecies were sequenced in this present study. Illumina sequencing of paired-end libraries provided approximately 10 Gb trimmed reads. The length of the complete cp-genomes ranged from 119,865 to 119,895 bp (Table 1). All cp-genomes had a quadripartite structure consisting of a pair of inverted repeat regions (IRs) (495 bp) separated by large single copy region(LSC) (65,630 to 65,691 bp) and small single copy region(SSC) (53,184 to 53,270 bp) regions (Fig. 1 and Table 1),which were similar to otherPinusspecies (Kang et al. 2019).Genome annotation identif ied a total of 108 genes, including 72 protein-coding genes, 36 tRNAs genes, and four rRNA genes. In these genes, 12 genes included one intron, and two genes contained two introns.
SSR analysis
The SSRs consist of 1-6 nucleotide repeat units and are widely used as molecular markers in population genetics (Rassi et al. 2002; Jeong et al. 2014). Among 69 SSRs regions identif ied here in the fourPinuscp-genomes, 17 were present inP. takahasii,P. densif loraandP. sylvestrisand 18 inP. funebris(Fig. 2), and 64 were composed only of A or T bases, four were composed of G bases, and one was composed of trinucleotide (TTA) repeats. No tetra-,penta- and hexanucleotide repeats were detected. In addition, among the 69 SSRs, 24 were located in the LSC region,45 were located in the SSC region, and none were in the IR region. Eight SSRs were protein-coding genes, and each species had two SSR locations inrpl32and inycf3. Poly(A) and poly(T) had been reported in a higher proportion to poly(G)and poly(C) in many plant families (Jeong et al. 2014; Konget al. 2017; Yu et al. 2020). The f indings of our study were consistent with those of another study on SSRs inPinuscp-genomes (Zeb et al. 2020), in whichP. sylvestrishad fewest SSR repeats among 97Pinuscp-genome sequences.Similarly in our study of four species,P. sylvestrishad the fewest SSRs in the cp-genome. The distribution of SSRs in gymnosperm cp-genomes is uneven (Ni et al. 2017a, b).Therefore, the diff erent types of SSRs, with their unique features, can be used as cp-markers. The SSRs identif ied in this present study provide a new perspective on the evolution and population genetics ofPinus(Ni et al. 2017a, b; Yu et al. 2020).
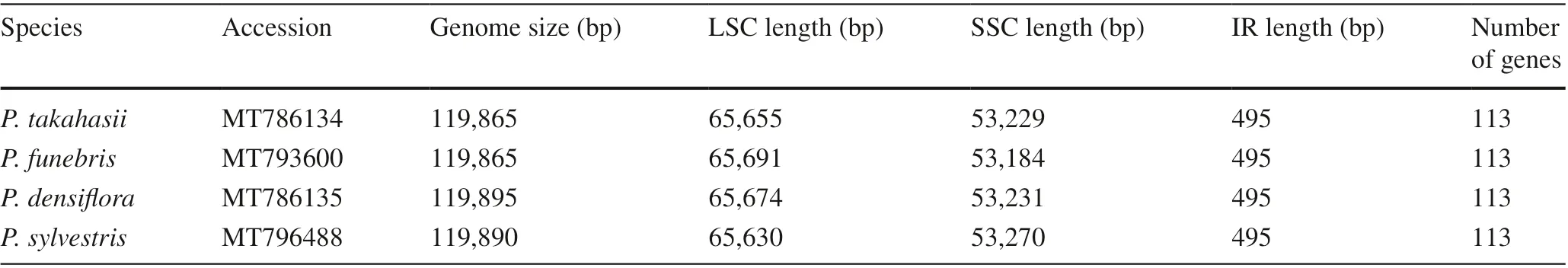
Table 1 Summary of complete chloroplast genomes for four Piuns species

Fig. 1 Complete chloroplast genome map of four Pinus species. Genes are color-coded based on the functional group. Genes inside the circle are transcribed clockwise; those outside are transcribed counterclockwise

Codon usage patterns in the chloroplast genomes of the four Pinus species
In total, the fourPinusspecies cp-genomes contained 20,089-20,117 codons. Among all the codons, leucine(Leu) was the most abundant amino acid with a frequency of 2013 (10.00%), 2011 (10.00%), 2010 (10.00%), and 2139(10.67%) inP. densif lora,P. funebris,P. sylvestris, andP.takahasii, respectively. Consistent with our study, other studies have shown that Leu was the most common codon in other land plant cp-genomes (He et al. 2017); it plays an important role in photosynthesis-related metabolism, and chloroplasts had a high demand for leucine biosynthesis(Park et al. 2017).
Our study also revealed synonymous codon usage bias;a high proportion of synonymous codons harbored an A or T(U) nucleotide in the third position. The preference for the high A - U (T) content at the third codon position is similar to various terrestrial plant cp-genomes (Raubeson et al. 2007; Liu et al. 2018). Furthermore, almost all gene A/U-ending codons had an RSCU value > 1, and C/G-ending codons had RSCU values < 1. It is generally believed that codon usage could ref lect the mode of gene mutation,so the RSCU values clustering results could provide useful information for phylogenetic relationship analysis (Nie et al. 2014).P. sylvestrisclustered withP. funebrisandP.takahasii, andP. densifl orawas in the outermost position,ref lecting the interpretation of fourPiunsphylogeny at the codon usage level (Fig. 3).
Boundary between LSC/SSC and IRs
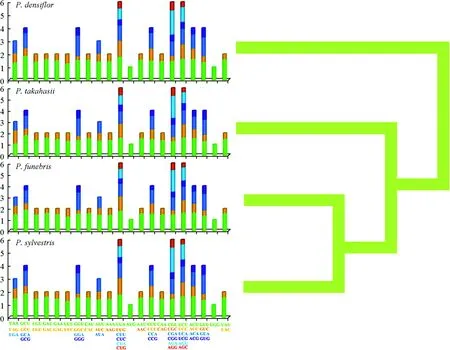
Fig. 3 Relative synonymous codon usages (RSCU) in protein-coding genes of four Pinus species. Codon families are indicated below the x-axis. Clustering was based on RSCU content shown on the right
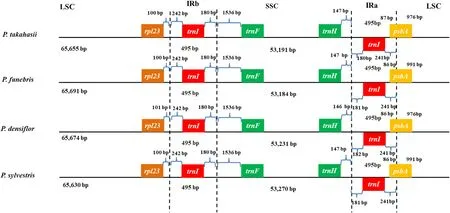
Fig. 4 Comparison of the boundary between LSC/SSC and IRs among four Pinus species. The numbers indicate the lengths of IGSs, genes, and spacers between IR-LSC and IR-SSC junctions
In gymnosperms, several species had been reported a loss of the large IR, especially inPinuscp-genomes, which had extremely shortened IR regions (Zeb et al. 2020). The LSC-IRb, SSC-IRb, SSC-IRa, and LSC-IRa junctions in the fourPinuscp-genomes were located betweenrpl23andtrnI,trnIandtrnF,trnHandtrnI, andtrnIandpsbA, respectively (Fig. 4). In cp-genomes, the expansion and contraction of IR regions are mainly responsible for structural variations in cp-genomes (Weng et al. 2017). The genomic region boundaries were highly similar and showed few base-pair displacements between the four species. Moreover, gene positioning at the IRs is more stable and conservative than for the LSC and SSC regions in the cp-genomes (Zeb et al.2020). No expansion or contraction events in IR regions were found among the fourPinusspecies, showing that the four species are closely related, without any structural changes in the cp-genomes. The LSC and SSC regions contributed to the observed diff erences in the overall size of the cp-genomes, and the situations were ref lected in genes adjacent to the border in the LSC and SSC regions with 1-63 bp variations. ThetrnF-GAAin SSC was located in 1535-1598 bp from the borders and had the most largest variation in length variation in this study.
Sequence divergence analysis in the four Pinus chloroplast genomes
Fig. 5 Sliding window analyses of aligned whole chloroplast genomes of four Pinus species. ( A) P. densif lora MT786135,P. sylvestris MT796488, P. takahasii MT786134, ( B) P. sylvestri s MT796488 and KR476379.1, ( C) P. densif lora (JN854210.1,MK285358.1, MF990371, MT786135). Window length 600 bp. Step size 200 bp
When the overall sequence identities of the fourPinuscpgenomes were plotted using DNAsp 5 withP. densif loraas the reference (Fig. 5), the coding regions were more conserved than the noncoding regions, and the f ive most highly divergent regions among the four cp-genomes all occurred in the noncoding regions. The f ive highly divergent regions were intergenic regionstrnE-clpP,cemA-ycf4, andpetDrpoAin the LSC andpsbD-trnTandtrnN-chlLin the SSC region. The intergenic regioncemA-ycf4was the most highly divergent region in species ofSalix(Chen et al. 2019).AlthoughpetD-rpoAandpsbD-trnThave been used for phylogenetic reconstruction of many plant groups (Fukuda et al. 2005; Minami et al. 2009; Byrne and Hankinson 2012;Tyler and J?nsson 2013), intergenic regionstrnE-clpPandtrnN-chlLhave never been used for phylogeographic or phylogenetic analyses of the genusPinus. In an earlier population genetic analysis ofPinusspecies (P. sylvestris,P. densif lora,P. funebrisandP. takahasii), the cpDNA sequences ofrpl16F71-rpl16R15,trnS-trnG,andrbcLdid not have high nucleotide variability (Ren et al. 2012). In future research of hybrid speciation inPinus, the observed divergence hotspot regions in this study could provide abundant information for marker development. The four highly divergent regions discovered inP. sylvestrisare shown in Fig. 5 B. Two (psbDtrnTandtrnN-chlL) were shown to be highly divergent in previous research onPinusspecies research, and the other two (psbBandrps8) were located in the coding regions.
Furthermore, gene sequence alignment results showed there were variations inpsbBmotifs, and positive selection analysis showed thatKa/Ks> 1. The genepsbBencodes a component of photosystem II in higher plants and is under strong positive selection in sun-loving rice species (Gao et al. 2019).Pinus, also a sun-loving species, may also have been subjected to strong selection pressure onpsbB, which could be treated as a molecular barcode for future research(Fig. S1 and Table S1). Inrps8, the sequence retained conserved motifs, andKa/Ks< 1 (Fig. S2 and Table S1).Compared to the fourP. densif loracp-genomes analyzed,that ofP. sylvestrishad an overall higher mutation rate.Five highly divergent regions are shown in Fig. 5 C. Aside from theycf3intron, which was unique toP. densif lora, the other four variable regions (trnE-clpP,petD-rpoA,psbD-trnT, andtrnN-chlL) were the same in the fourPinusspecies.
Phylogenomic reconstruction based on whole plastomes
The phylogenetic reconstruction was based on 30 whole cp-genome sequences of species belonging to Pinaceae,Abies, and Taxaceae. The ML phylogenetic results resolved 27 nodes with bootstrap support values of 46-100%, 12 of which were 100% (Fig. 6). This topology was in accordance with previous phylogenetic analyses that used cp-proteincoding regions (Gernandt et al. 2005; Kang et al. 2019).P.sylvestris,P. densifl ora,P. funebrisandP. takahasiiclustered into one group, the same as the codon usage clustering result (Fig. 6). Studies have shown that the closer the genetic relationship the more similar the codon usage pattern will be (Tang et al. 2021). Thus, there was a correlation between the codon usage bias of chloroplast genes and the genetic relationship withinPinusspecies. Furthermore,P. funebrisandP. takahasiiwere related toP. sylvestris. Cp-genomesare inherited paternally in Pinaceae species, which may indicate thatP. sylvestrisis the paternal parent ofP. funebrisandP. takahasii. The f indings of the current study are consistent with previous f indings based on a population genetic analysis using chloroplast, mitochondrial, and nuclear sequences thatP. densif loraserved as the maternal parent ofP. funebrisandP. takahasii(Ren et al. 2012). Since the paternal parents of these hybrid species might not have been included in our present study, species sampling should be increased in future population genetics studies.

Studies on the biogeography and phylogeny ofPinususing mitochondrial and cpDNA markers have revealed the structure and reconstructed the phylogeny (Wu et al. 2020;Zaborowska et al. 2020). This comparative analysis of the cp-genomes of the endangeredAcer miaotaiensefrom f ive ecological regions in China and phylogenetic reconstruction deepened our understanding of the impact of diff erent environments on genetic diversity (Zhao et al. 2018).
The cp-genomes forP. densifl oraused in the present study were from trees in Shangdong Province clustered with a Korean sample (MF990371.1). Our results showed that the genetic diversity among diff erent populations ofP. densif lorawas low, which may be due to the extensive dissemination of gymnosperm pollen and the promotion of gene exchange (Ran et al. 2015). Our results showed a lower amount of intraspecif ic variation among the cp-genomes ofP. densif lora(Fig. 5 C). Some diff erences were also identif ied in diff erent phylogenetic analyses in whichP. kesiya,P.yunnanensisandP. densataclustered into diff erent groups(Fig. 6 ; Fig. S3). Previous research has shown these species can be confused and were their phylogenetic resolution was mostly low (Zeb et al. 2020). Taken together, our results using the cp-genome provides insights into the phylogenetic relationship among fourPinusspecies in Northeast China.In the future, more detailed taxon sampling will contribute more comprehensively to the phylogenetic study ofPinus.
Conclusions
In the present study, we sequenced and analyzed complete plastomes of fourPinusspecies, providing new insight into homoploid hybrid speciation and phylogenetic relationships. Through comparison, we found that the cp-genomes of the four species were similar in structural characteristics and had a typical four-part structure.Phylogenomic analyses based on cp-genome sequences yielded robust relationships withinPinus, which indicate that the cp-phylogenomic approach could be employed to tackle intractable phylogenies. In total, 14 highly variable loci were chosen as potential molecular markers for the fourPinusspecies, which could use in population genetic studies. In sum, these data and results presented here will enhance future utilization for these valuablePinusspecies.
Funding This research was funded by Kunyu Mountain National Nature Reserve Administration.
Declarations Conf lict of interest The authors declare that they have no conf lict of interest.
 Journal of Forestry Research2022年6期
Journal of Forestry Research2022年6期
- Journal of Forestry Research的其它文章
- Mixing planting proportions in a plantation aff ects functional traits and biomass allocation of Cunninghamia lanceolata and Phoebe bournei seedlings
- Intraspecif ic leaf morphological variation in Quercus dentata Thunb.: a comparison of traditional and geometric morphometric methods, a pilot study
- Mapping the potential distribution suitability of 16 tree species under climate change in northeastern China using Maxent modelling
- Correction to: Eff ects of active molecules of Korean pine seed on rodent health and implications for forest regeneration
- Seed, germination, and seed-reserve traits diff er along an altitudinal gradient
- Genetic variation and superior provenances selection for wood properties of Larix olgensis at four trials