
Role of apoptosis-inducing factor in perinatal hypoxic-ischemic brain injury
2021-09-01 05:43,,,,,
中國神經(jīng)再生研究(英文版) 2021年2期
, , , , ,
Abstract Perinatal complications, such as asphyxia, can cause brain injuries that are often associated with subsequent neurological de ficits, such as cerebral palsy or mental retardation. The mechanisms of perinatal brain injury are not fully understood, but mitochondria play a prominent role not only due to their central function in metabolism but also because many proteins with apoptosis-related functions are located in the mitochondrion. Among these proteins, apoptosis-inducing factor has already been shown to be an important factor involved in neuronal cell death upon hypoxia-ischemia, but a better understanding of the mechanisms behind these processes is required for the development of more effective treatments during the early stages of perinatal brain injury. In this review, we focus on the molecular mechanisms of hypoxic-ischemic encephalopathy, speci fically on the importance of apoptosis-inducing factor. The relevance of apoptosis-inducing factor is based not only because it participates in the caspase-independent apoptotic pathway but also because it plays a crucial role in mitochondrial energetic functionality, especially with regard to the maintenance of electron transport during oxidative phosphorylation and in oxidative stress, acting as a free radical scavenger. We also discuss all the different apoptosis-inducing factor isoforms discovered, focusing especially on apoptosis-inducing factor 2, which is only expressed in the brain and the functions of which are starting now to be clari fied. Finally, we summarized the interaction of apoptosis-inducing factor with several proteins that are crucial for both apoptosis-inducing factor functions (prosurvival and pro-apoptotic) and that are highly important in order to develop promising therapeutic targets for improving outcomes after perinatal brain injury.
Key Words: apoptosis; apoptosis inducing factor; asphyxia; cell death; free radical;hypoxia-ischemia; mitochondria; neonates; oxidative stress
Introduction
Perinatal brain injury is a major public health issue and is a leading cause of neonatal mortality and morbidity (Liu et al., 2016). It may lead to developmental impairment and permanent neurological deficits, such as cerebral palsy and mental retardation (Lally et al., 2019; Juul et al., 2020). Among many etiological factors, hypoxic-ischemic encephalopathy(HIE) in term infants and intraventricular/periventricular hemorrhage in preterm infants are the most common causes of perinatal brain damage (Hagberg et al., 2015). The different etiological factors during critical developmental periods can lead to a common pathway of perinatal brain injury marked by neuronal excitotoxicity, cellular apoptosis, and microglial activation (Hagberg et al., 2014; Wu et al., 2019). Apoptosis has been found more prominent in the immature brain compared to the mature brain (Zhu et al., 2005). Some clinical studies have shown promising results for perinatal brain injury(Zhu et al., 2009a; Azzopardi et al., 2016); however, widely accepted clinically efficient therapy is still limited (Tagin et al.,2015; Hagberg et al., 2016). Thus, there is a pressing need for a better understanding of the mechanisms of neuronal cell death and perinatal brain injury and for conducting comparative and translational studies on how to reduce neuronal cell death, increase cell survival, and promote brain regeneration and repair after injury. Perinatal hypoxiaischemia (HI)-induced brain injury is one of the most common forms of neonatal brain injury, which is more common in the developing countries (Liu et al., 2016). Thus, we focus on the molecular mechanisms of hypoxic-ischemic brain injury in this review, speci fically the importance of apoptosis-inducing factor (AIF) as a key protein for finding new therapeutic strategies for preventing perinatal brain injury.
An electronic search of the Google Scholar and PubMed for literature describing apoptosis-inducing factor in perinatal hypoxic-ischemic brain injury from 1996 to 2020 was performed using the following conditions: (AIF [All Fields] OR “apoptosisinducing factor”[All Fields]) NOT (“neoplasms”[MeSH Terms]OR “neoplasms”[All Fields] OR “cancer”[All Fields]).
Clinical Consequence of Hypoxic-Ischemic Encephalopathy
HIE is a condition that is associated with oxygen deprivation in the neonate due to perinatal asphyxia. Despite important progress in neonatal care over the last decades, HIE is still an important contributor to neonatal mortality and to major neurodevelopmental disabilities, including mental retardation,cerebral palsy, learning disabilities, and seizures (Van Handel et al., 2007; Douglas-Escobar and Weiss, 2015; Y?ld?z et al.,2017; Schreglmann et al., 2020). Due to improvements in perinatal care and the increased survival rate of infants born with a low gestational age, the absolute number of subjects affected by these complications has increased. The incidence of HIE ranges from 1 to 8 per 1000 live births in developed countries, but underdeveloped countries have reported incidences of up to 26 per 1000 live births (Hayakawa et al.,2014; Douglas-Escobar and Weiss, 2015). Hypoxic-ischemic brain injury can occur in newborns at any gestational age,but preterm and very preterm babies are less prepared to adapt to perinatal insults compared to term infants, making them more vulnerable to neurodevelopmental impairments due to hypoxia-ischemia (HI; Gopagondanahalli et al.,2016). Although premature babies are able to tolerate more prolonged periods of HI, the effect can dramatically alter the normal development of the immature brain (Bennet et al.,2013; Galinsky et al., 2018). HIE can be ranked in three clinical stages according to the widely accepted Sarnat scoring system as mild, moderate, and severe encephalopathy (Sarnat and Sarnat, 1976). The few infants who survive severe HIE will most likely develop life-long complications (Hankins and Speer,2003), while for moderate HIE, some estimations suggest that about 30-40% of the survivors develop cerebral palsy or other major neurological impairments and another 30-40%develop cognitive problems (Lindstr?m et al., 2006). Other studies have shown that untreated moderate to severe HIE results in an approximately 60% risk of either death or major neurological impairments, which is a major burden to those afflicted, their families, and society (Carli et al., 2004; Edwards et al., 2010; Natarajan et al., 2016).
Mechanisms of Hypoxic-Ischemic Brain Injury
Asphyxia in newborn infants causes brain injury that lasts for several days due to mechanisms of cellular apoptosis,autophagy, necrosis, and inflammation, and mitochondria play extremely important roles in these processes (Xie et al.,2016; Hua et al., 2017; Thornton et al., 2017; Li et al., 2019).The type of cell death depends on several factors, including the severity of the insult, the cell type, the metabolic stress,the sex of the infant, and the elapsed time since the event(Demarest et al., 2016; Thornton et al., 2017; Charriaut-Marlangue et al., 2018). Apoptosis is a physiological process during normal brain development, whereby cells are discretely removed. Studies have shown that apoptotic cell death machinery is activated in pathological conditions, such as HI,and apoptosis is much more prominent in the immature brain.
Apoptosis
Two main apoptotic pathways occur in the cell: the extrinsic pathway mediated by death receptors and the intrinsic pathway mediated by the mitochondria (Thornton et al.,2017). Both routes take place through multiple molecular mechanisms and are related to each other, such that the molecular events of one in fluence the other. Contrary to the extrinsic pathway (which is triggered by extracellular signals that oligomerize death receptors located on the plasma membrane (Wajant, 2002)), the intrinsic pathway is triggered by intracellular stimuli, such as an increase of reactive oxygen species, nitric oxide, and Ca2+caused by glutamate overflow (Hagberg et al., 2014; Sutcliffe et al., 2017). The extrinsic pathway activates BID protein (a pro-apoptotic B-cell lymphoma 2, BCL-2 family member, also known as BH3 interacting-domain death agonist), which in turn oligomerize BCL-2-associated X protein and BCL-2 homologous antagonist killer 1 (BAK1) (Korsmeyer et al., 2000; Wang et al., 2010).These proteins play a crucial role in mitochondrial outer membrane permeabilization (MOMP) and pore formation(Zhu et al., 2010; Shamas-Din et al., 2011; Green and Llambi,2015; Stehle et al., 2018). Subsequently, the release from the mitochondria to the cytosol of pro-apoptotic proteins will occur, including cytochrome c (Cyt c), AIF, second mitochondria-derived activator of caspases, and endonuclease G (Thornton et al., 2017), making this a crucial event for another way to classify apoptosis, depending on the final executor, and that differ in their dependence or independence on cysteine-aspartic proteases (caspases). Caspase-dependent apoptosis is initiated by Cyt c and the subsequent assembly of the apoptosome complex in the cytosol. This event will lead to the activation of caspase 3, which will cut the DNA into small pieces of about 200 to 1000 bp (J?nicke et al.,1998). The caspase-independent pathway terminates with the translocation of AIF (also known as “parthanatos”) to the nucleus mainly, where it will cause large-scale DNA fragmentation (into pieces of about 50,000 bp) and chromatin condensation (Galluzzi et al., 2009; Fatokun et al., 2014).
Apoptosis is a crucial process in the immature brain, and it determines the appropriate development of the central nervous system. Unsurprisingly, many components of the intrinsic pathway, as well as proteins involved in apoptosis(such as caspase 3, BCL-2 family proteins, and AIF) are upregulated during brain development (Thornton et al.,2017). Indeed, these apoptotic mechanisms, including nuclear translocation of AIF, Cyt c release, and caspase 3 activation,have been found to be more prominent in immature than in juvenile and adult mouse brains (Zhu et al., 2005).
Apoptosis-inducing factor
AIF (full gene name: apoptosis-inducing factor, mitochondriaassociated 1; AIFM1) is a flavin adenine dinucleotide(FAD)-dependent, nicotinamide adenine dinucleotide(NAD+) oxidoreductase that is located in the mitochondrial intermembrane space under physiological conditions (Candé et al., 2002). AIF is a highly conserved protein, and it has significant homology with the oxidoreductases of bacteria,plants, and fungi (Elguindy and Nakamaru-Ogiso, 2015).In humans, the gene encoding AIF is located at the q26.1 region of the X chromosome. The most abundant transcript,calledAIFM1(Gene ID: 9131), possesses a transcribed region encoded in 16 exons that generates a 67 kDa precursor molecule of 613 amino acids (AAs) in humans (Susin et al.,1999b). AIF1 has three domains: FAD-bipartite binding domain(AAs 129-262 and AAs 401-480), a NADH-binding motif (AAs 263-400), and a C-terminal domain (AAs 481-608) (Lorenzo et al., 1999). In addition, there are two mitochondrial localization sequences (MLSs) placed one after the other in the N-terminal region (AAs 1-41) and two nuclear localization sequences(NLS1 and NLS2) located within the two FAD-binding domains(Porter and Urbano, 2006). The MLS drives the transportation of AIF into the mitochondrial intermembrane space (Susin et al., 1996; Zamzami et al., 1996). Once there, AIF will undergo the first proteolytic cleavage and thus be processed to a mature form (62 kDa) (Figure 1). AIF is then inserted through its amino-terminal transmembrane segment into the inner mitochondrial membrane (IMM), leaving the rest of the protein exposed to the mitochondrial intermembrane space(Otera et al., 2005). The amino-terminal transmembrane domain (TMD) encompasses AAs 54 (where the mitochondrialprocessing peptidase cleavage site is located) to 103 (where the calpain/cathepsin cleavage site is located). Within the amino-terminal transmembrane segment is the TMD (AAs 67-83). AIF also has two binding domains: one for heat shock protein 70 (HSP70) at AAs 150-228 (included in the FAD-binding domain), another for cyclophilin A (CypA) at AAs 367-399 (included in the NADH-binding motif), and the last one for macrophage migration inhibitory factor (MIF) at AAs 567-592. AIF also possesses two DNA-binding sites located in AAs 255-265 and 510-518 (Figure 1).
In the absence of apoptotic stimuli, AIF is essential for obtaining energy in the mitochondria, as well as for the optimal functioning of the respiratory chain (Vahsen et al.,2004), making AIF an essential protein for survival (Joza et al.,2001). AIF involvement in free radical formation and removal has also been suggested (Klein et al., 2002; Ishimura et al.,2008). In addition, some studies have suggested a relationship between AIF’s redox function with the biogenesis and/or assembly of complexes I and III of the electron transport chain during oxidative phosphorylation (OxPhos), as well as with the maintenance of these complexes for optimal performance(Fato et al., 2008; Hangen et al., 2010a). Interestingly,in vitrostudies have shown that AIF depletion by AIF siRNA in a model system of glutamate toxicity in immortalized hippocampal HT-22 neurons had effects similar to rotenone,a speci fic mitochondrial complex I inhibitor, thus mediating a preconditioning effect at the level of the mitochondria (?xler et al., 2012).
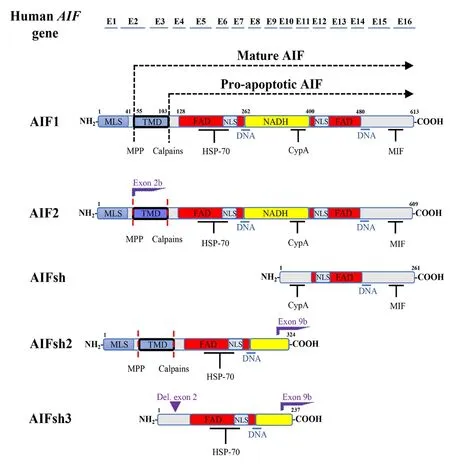
Figure 1 | Schematic representation of the different human AIF isoforms and their binding domains.
In the presence of apoptotic stimuli, AIF plays a central role in neuronal cell death (Kroemer et al., 2007; Hagberg et al.,2014). In 2003, we found that AIF was involved in neuronal cell death after HI in the neonatal rat brain (Zhu et al., 2003). AIF was detected in the nuclei just a few hours after HI and only in damaged areas. Furthermore, the larger the infarct volume Shown are the exons of theAIFgene (E1-E16) and the N-terminal, MLS, TMD,FAD, NLS, NADH and C-terminal domains, together with the HSP70, DNA,MIF and CypA biding sites of the different AIF protein forms. Mitochondrialprocessing peptidase and calpains cleavage sites are also revealed, generating the mature AIF and the pro-apoptotic AIF forms, respectively. The difference in the color intensity of the TMD domain of AIF2 re flects the alternative usage of exon 2b, affecting the TMD segment. AIFsh, AIFsh2, and AIFsh3 are generated by the alternative splicing of exon 9b. AIFsh3 has a structure similar to AIFsh2 but with the deletion of exon 2, which leads to the loss of MLS and TMD domains. AIF: Apoptosis-inducing factor; CypA: cyclophilin A; FAD: flavin adenine dinucleotide; HSP70: heat shock protein 70; MIF: migration inhibitory factor; MLS: mitochondrial localization sequence; MPP: mitochondrialprocessing peptidase; NADH: nicotinamide adenine dinucleotide; NLS: nuclear localization sequences; TMD: transmembrane domain.was, the greater the number of AIF-positive nuclei were found and treating the mice with a multi-caspase inhibitor did not alter the number of AIF-positive nuclei (Susin et al., 1999a;Zhu et al., 2003). We know now that, after HI, AIF is cleaved from the IMM by calpains and/or cathepsins in its apoptotic form (Figure 1) (Polster et al., 2005; Yuste et al., 2005), with a molecular weight of 57 kDa (Otera et al., 2005). The protein refolds while incorporating FAD and is then released into the cytosol, where it combines with CypA. After this, the complex translocates to the nucleus (Zhu et al., 2007a). Artus et al.(2010) subsequently found that, once in the nucleus, AIF also interacts with H2A histone family member X (H2AX). H2AX is linked to DNA damage repair by modifying the chromatin structure and making damaged DNA sites accessible to repair factors (Paull et al., 2000). The AIF/H2AX complex improves DNA’s accessibility to AIF (Cande et al., 2004), thus making the synchronized presence of these three proteins a team with specialized roles: AIF (binding domains), H2AX(generation of DNA accessibility), and CypA (DNase activity)(Artus et al., 2010). Therefore, this set of molecules in the nucleus acts as a DNA degradation complex and is required for large-scale DNA fragmentation and chromatinolysis,which ultimately leads to cell death in a caspase-independent manner. Chromatinolysis mediated by H2AX has also been observed in caspase-dependent apoptosis, in which the DNA degradation is of a different nature, and this could be because of the different actors implicated: caspase 3/caspase-activated DNase or AIF/CypA, respectively (Artus et al., 2010). There is some controversy regarding how the interaction between AIF and CypA enables their translocation to the nucleus(Doti and Ruvo, 2017). One model suggests the independent translocation of both proteins prior to chromatinolysis and DNA fragmentation, and this is supported by the results in a necrosis model in which CypA down-regulation did not compromise the nuclear translocation of AIF but did reduce the extent of DNA damage (Artus et al., 2010). The other model proposes that, upon apoptotic stimuli, AIF and CypA form a complex in the cytosol, and this interaction allows both proteins to translocate to the nucleus and induce cell death (Cande et al., 2004; Zhu et al., 2007a; Doti et al., 2014;Figure 2).
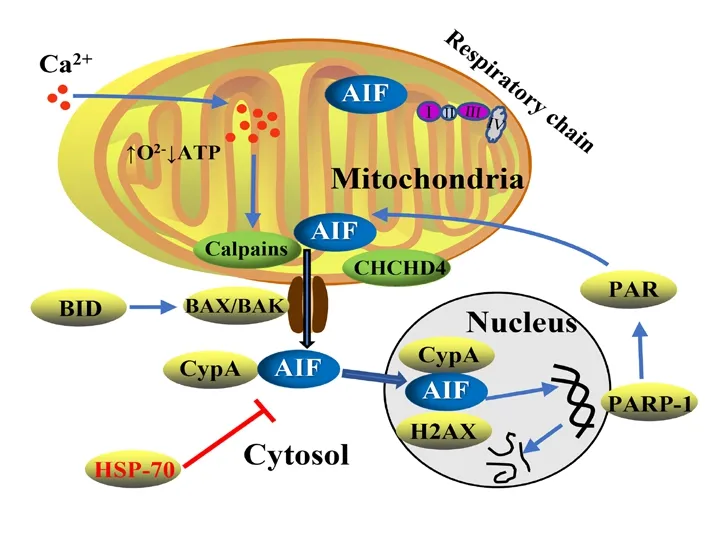
Figure 2 | Schematic representation of the AIF translocation from the mitochondria to the nucleus upon apoptotic stimuli and its regulation by other proteins.
Calpains will cleave AIF from the IMM. BCL-2 pro-apoptotic protein BID,among others, will promote the MOMP and pore formation. PAR polymers(produced by the overactivation of PARP-1) and CHCHD4 protein are also involved in the translocation of AIF from the mitochondria. Once in the cytosol, AIF will bind to CypA. The HSP70 chaperone can neutralize AIF in the cytosol. In the nucleus, the AIF/CypA complex will cooperate with H2AX to induce chromatin condensation and large-scale DNA fragmentation.AIF: Apoptosis-inducing factor; ATP: adenosine triphosphate; BAK: BCL-2 homologous antagonist killer; BAX: BCL-2-associated X; BID: BH3 interactingdomain death agonist; CHCHD4: coiled-coil-helix-coiled-coil-helix domaincontaining protein 4; CypA: cyclophilin A; H2AX: H2A histone family member X; HSP70: heat shock protein 70; PAR: poly-ADP ribose; PARP-1: poly (ADP-ribose) polymerase 1.
Apoptosis-inducing factor isoforms
Recent studies have shown the existence of more variants of AIF (Figure 1). AIF2 contains 609 AAs (608 AAs in mice), and it expresses an alternative exon (2b) compared to the other isoform (Loeffler et al., 2001). In opposition to AIF1 (which is ubiquitously expressed), AIF2 is only expressed in the brain(Hangen et al., 2010b). It has been shown that AIF2 is also found in the IMM, but as a result of the alternative splicing of exon 2 (which introduces a short difference in the second MLS of the protein), the segment that is used to anchor the protein into the IMM does so more firmly due to changes in the hydrophobicity of the AAs compared to the other isoform(Hangen et al., 2010b). AIF has been shown to be a major contributor to caspase-independent neuronal loss induced by neonatal cerebral HI, and the Harlequin (Hq) mice, which have an 80% reduction in the expression of the protein (including both isoforms AIF1 and AIF2), showed an impressive reduction of brain injury after HI (Zhu et al., 2007b). However, the brainspecific isoform AIF2 has barely been studied. Previously,one of the few studies on AIF2 showed that, in humans, the mRNA expression ofAIFM2was higher in the adult brain than in the fetal brain, while the mRNA expression ofAIF1was lower (Hangen et al., 2010b). As explained before, the AIF2 isoform is only expressed in the brain, but Hangen and colleagues showed that its distribution is not homogeneous.Approximately 25% of the cells present in the anterior olfactory nucleus stained uniquely for the exon 2b-specific probe, indicating the existence of brain cells that express AIF2 but not AIF1. In that paper, it was also mentioned that AIF2 was more difficult to dissociate from the mitochondrial membrane than AIF1 due to the changes in the AA sequences that affect the region of the protein that is anchored in the IMM, suggesting that AIF2 might contribute less efficiently to apoptosis than AIF1 (Hangen et al., 2010b). AIF has the ability to form dimers or higher-order oligomers, and these complexes can potentially be formed by both AIF1 and/or AIF2 isoforms. Upon apoptotic stimuli, the heterodimers or heterooligomers containing one or more AIF2 isoforms would be less likely to be released into the cytosol. Therefore, the AIF1/AIF2 ratio in the IMM might be crucial for the pro-apoptotic role of AIF.
The results in a recentAif2knock out (KO) study (Rodriguez et al., 2018) showed that by eliminating the expression of AIF2, AIF1 will increase its expression under physiological conditions by 38% in 9-day-old mice and by 90% in 60-dayold mice as a compensatory reaction. This increase together with the fact that the AIF2 isoform is not expressed, increases the AIF1/AIF2 ratio substantially. In the wild-type mice from this study, under physiological conditions, a natural increase inAif2mRNA expression with age (64% higher) was observed,while Aif1 expression remained stable (Rodriguez et al., 2018).AIF is essential for embryonic development, and without it,the programmed cell death necessary during cavitation of embryoid bodies does not occur (Joza et al., 2001). WhileAifKO mice are embryonically lethal, Hq mice are viable but display severe phenotypes (Klein et al., 2002). However, in theAif2KO study, it was found that AIF2 was not essential for mouse viability, and no phenotype was observed inAif2KO adult mice under physiological conditions, except an increase of neurogenesis in the sub-granular zone of the dentate gyrus of the hippocampus in 60-day-old mice (Rodriguez et al.,2018). Interestingly, Hq mice showed progressive loss of adult cerebellar and retinal neurons, and in experiments withAifKO embryos, the mutant mice had midbrain defects and dramatic deficits in cerebellar Purkinje and granule cell precursors(Ishimura et al., 2008).AifKO mice displayed greater brain injury at 72 hours after HI, which was attributed to an increase in the expression level of AIF1 whenAifm2was knocked out.This induced a change in the AIF1/AIF2 ratio that promoted increased oxidative stress and subsequent brain injury after HI(Rodriguez et al., 2018).Other types of AIF isoforms have also been discovered. AIFsh is produced from an alternative origin of transcription located at intron 9 of theAIFgene. As a result, AIFsh contains the C-terminal AIF domain encoded by exons 10 to 16, lacking MLS, and therefore cannot translocate to the mitochondria.AIFsh is a cytosolic protein that causes the same apoptotic effects as AIF1 but lacks oxidoreductase function. AIFsh is ubiquitously expressed (like AIF1), but a different expression pattern was observed through the tissues between AIF1 and AIFsh (Delettre et al., 2006a). Two other human isoforms have also been discovered: AIFsh2 and AIFsh3 (Delettre et al., 2006b), which are generated by alternative splicing of the exon 9b. AIFsh2 contains AAs 1-324, while AIFsh3 is formed by AAs 87-324, meaning that both lack the C-terminal and NLS2 domains; therefore, they are not translocated to the nucleus (and they both lack the proapoptotic function of AIF).In addition, AIFsh3 also lacks the MLS sequence due to the splicing of exon 2; therefore, the protein is not translocated to the mitochondria (Figure 1). Interestingly, AIFsh2 is absent in brain tissue (Delettre et al., 2006b).
Importance of apoptosis-inducing factor in perinatal hypoxic-ischemic encephalopathy
The importance of AIF in neuronal cell death after HI was first noticed by using wide spectrum caspase inhibitor, which did not affect AIF release and DNA damage. Furthermore, the number of AIF-positive nuclei was positively correlated with brain injury (Zhu et al., 2003). This was further con firmed by using 9-day-old Hq mice, in which the level of expression of Aif was drastically reduced to 20% of the levels found in wildtype (WT) mice (Zhu et al., 2007b). Hq mice were originally observed to undergo ataxia due to cerebellar atrophy and blindness due to retinal degeneration (Klein et al., 2002). In Hq mice exposed to HI insult, the infarct volume after HI was reduced by 53% in male (YXHq) mice and by 43% in female(XHqXHq) mice. This also confirmed the independence of AIF from the caspase-dependent apoptosis pathway, because the Hq mutation did not inhibit the release of Cyt c after HI, nor the activation of caspase 3, even though Hq mice displayed half the injury severity compared to WT mice. Furthermore,the combined protective effect of the Hq mutation, together with the administration of a caspase inhibition, reduced the infarct volume more than 75%, showing that AIF and caspase act in parallel (Zhu et al., 2007b). Hq mouse neurons were found to be particularly sensitive to oxidative stressinduced cell death, which suggested that AIF indeed acts as a mitochondrial scavenger of reactive oxygen species and/or that AIF is involved in OxPhos. Neonatal Hq mice also exhibited 18% less respiratory chain complex I and 30%less catalase compared with WT mice. Finally, when the Hq mice were administrated an anti-oxidant agent (edaravone),the infarct volume was even more reduced compared to the reduction seen in Hq mice alone (~75% reductionvs.~50% reduction) and was much more reduced than when edaravone was administrated to WT mice (~20% reduction in infarct volume). This was attributed to the fact that edaravone restored the anti-oxidant defense compromised by the Hq mutation (Zhu et al., 2007b).
Our recent study, by using overexpressing AIF1 transgenic mice (without affecting the expression of AIF2), showed increased brain injury after HI compared to WT littermates(Li et al., 2020). Similar to the results shown in the AIF2 KO study (Rodriguez et al., 2018), under physiological conditions,the overexpression of Aif1 also promoted neurogenesis in the sub-granular zone of the dentate gyrus in adult mice, with no other phenotypic changes. Both studies led us to believe that the increase in the expression of AIF1 (either by knocking outAif2or by knocking in a pro-viral insertion ofAif1) was behind this observed neurogenesis.
Regulation of apoptosis-inducing factor
Different colors and symbols were used based on the kind of event: general biological information discovered (black diamond), new isoform discovered(blue triangle), and new protein interaction discovered (red star 8 point).AAs: Amino acids; AIF: apoptosis-inducing factor; BCL-2: B-cell lymphoma 2; CHCHD4: coiled-coil-helix-coiled-coil-helix domain-containing protein 4;CypA: cyclophilin A; Cyt c: cytochrome c; Hq: harlequin; H2AX: H2A histone family member X; HSP70: heat shock protein 70; IMS: intermembrane space; KO: knock-out; MIF: migration inhibitory factor; MIMS: mitochondrial intermembrane space; MLS: mitochondrial localization sequence; PAR: poly-ADP ribose; RIP3: receptor-interacting protein 3; ROS: reactive oxygen species;TRX: thioredoxin.
Protein interaction:The AIF switch from a pro-survival to a pro-apoptotic protein is controlled by different mechanisms.The first mechanism involves MOMP, which is regulated by several proteins and serves as a good example of how the caspase-dependent and caspase-independent pathways are related to each other. The extrinsic pathway activates caspase 8, which in turn can activate the protein BID, that,as mentioned before, will lead to the MOMP and pore formation. As a consequence, cell death-related proteins will be released to the cytosol, belonging to both caspasedependent and caspase-independent pathways (Green and Llambi, 2015). Second, AIF is regulated by the activation of calpain and cathepsin proteases, which are needed to cleave AIF from the IMM (Polster et al., 2005; Cao et al.,2007). Third, AIF’s apoptotic function is controlled by the HSP70 chaperone, which can neutralize AIF by binding to the specific HSP70-binding domain located in AAs 150-228 of mature AIF (Ravagnan et al., 2001). Lastly, AIF is also regulated by poly (ADP-ribose) polymerase 1 (PARP-1), which is a nuclear enzyme involved in DNA repair, by producing poly-ADP ribose polymers using NAD+However, in case of excessive DNA damage caused by severe genomic stress,overactivation of PARP-1 can drain the cellular NAD+supply and cause energy depletion (Gill and Perez-Polo, 2008). PARP-1 has been suggested to be a key contributor to cell death,and indeed, the term parthanatos is being used now to refer to the caspase-independent apoptotic pathway based on the overactivation of PARP-1 and consequent NAD+depletion(David et al., 2009). In addition, PARP-1 overactivation participates in the translocation of AIF from the mitochondria to the nucleus, and in aPARP-1KO study in mice, neurons failed to release AIF to the nucleus in response to apoptotic stimuli (Yu et al., 2002;Figure 2).
It has been established that AIF’s pro-survival function requires a functional coiled-coil-helix-coiled-coil-helix domain-containing protein 4 (CHCHD4) protein, andAifdepletion is correlated with decreased levels of CHCHD4 protein (without affecting mRNA transcription) (Hangen et al., 2015). Furthermore, high levels of CHCHD4 protein have been shown to help offset the problems associated with low levels of AIF, and low levels of CHCHD4 protein have also been shown to lead to similar problems as low levels of AIF(Hangen et al., 2015; Meyer et al., 2015). All of these results suggest that both proteins work epistatically, at least in terms of mitochondrial regulation. In aChchd4-happloinsufficiency study (Sun et al., 2017), it was shown that reducing the protein expression of theChchd4gene by 25% reduced brain damage in 9-day-old mice after HI but did not affect AIF protein expression, suggesting that CHCHD4 protein acts by blocking the release of AIF from the mitochondria upon apoptotic stimuli (as well as blocking Cyt c release;Figure 2).Other proteins have been suggested to interact with AIF. The thioredoxin system is one major redox system in mammalian cells and cytosolic thioredoxin-1 interacts with AIF in physiological conditions, but this interaction is disrupted under pro-apoptotic oxidative stress conditions. Importantly, AIF-mediated DNA damage was attenuated when the expression of thioredoxin-1 was reduced (Shelar et al., 2015). MIF has also been suggested to interact with cytosolic AIF before both of them are translocated to the nucleus. MIF would act as an endonuclease that contributes to large DNA fragmentation induced by PARP-1 (Wang et al., 2016). Lastly, a cytosolic interaction between AIF and receptor-interacting protein 3 in neuronal programmed necrosis has been proposed, with their subsequent translocation to the nucleus inducing necroptosis(Xu et al., 2016). The breakthrough discoveries about AIF have been summarized in a timeline from the first evidence of AIF in 1996 until now (Figure 3).
Oxidative stress:Free radicals are formed in the brain as a
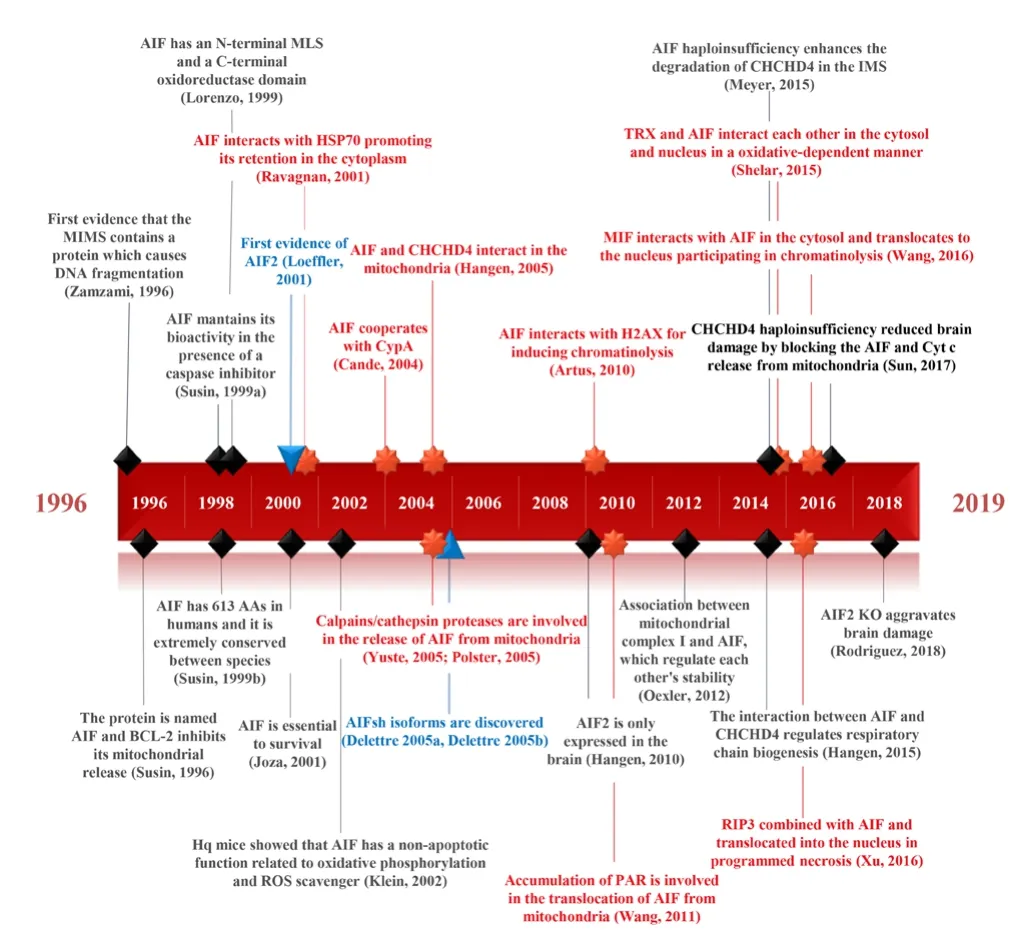
Figure 3|Timeline with the breakthrough discoveries about AIF.
consequence of the HI insult and reperfusion process, and they are implicated in the development of perinatal brain injury (B?genholm et al., 1997; Ferriero, 2001; Wang et al.,2007). The excessive production of free radicals will lead to oxidative stress that will affect different macromolecules,including lipids, proteins, and nucleic acids (Li et al., 2019).The accumulation of free radicals plays an important role in apoptosis, by modulating mitochondrial permeabilization, and the release of mitochondrial intermembrane pro-apoptotic proteins to the cytosol (Galluzzi et al., 2009; Hagberg et al., 2014). Mitochondria, especially mitochondria in brain cells, are also crucial in the development of oxidative stress,because their energy demands are high. Thus, they are a major source of free radical production (Sch?nfeld and Reiser,2013). The consequences of such stress will negatively affect the metabolic processes taking place in mitochondria, leading to a decrease in ATP production (Sun et al., 2016).
AIF also plays a crucial role in mitochondrial energetic functionality, especially with regard to the maintenance of electron transport chain complexes I and III during OxPhos(?xler et al., 2012). The AIF oxidase-reductase activity was suggested byin vitroexperiments performed using natural AIF puri fied from mitochondria that exhibited NADH oxidase activity (Miramar et al., 2001; Hong et al., 2015). This activity was shown to be independent from its role in apoptosis because of a recombinant AIF form lacking the FAD domain;therefore, its NADH oxidase activity did not lose its apoptotic function (Miramar et al., 2001). In addition, the AIFsh isoform showed apoptotic activity, despite lacking oxide-reductase activity (Delettre et al., 2006a), while AIFsh2 retained oxidereductase activity, despite lacking apoptotic activity (Delettre et al., 2006b). A similar experiment was replicated some years later, suggesting that AIF is a redox-signaling molecule in which its pro-survival and its pro-apoptotic roles are controlled by NADH (Churbanova and Sevrioukova, 2008). Furthermore, it was pointed out that NAD+reduction would cause a transition in AIF protein from a monomeric to a dimeric form; moreover,
AIF dimerization could potentially lead to a conformational change in the protein affecting the accessibility of the NLS2 binding domain and DNA binding sites (Sevrioukova, 2009).
This property of AIF to form dimers also plays an important role when it comes to explaining some of the results obtained inAif2KO study (Hangen et al., 2010b; Rodriguez et al.,2018). The hypothesis behind this is based on the potential capability of AIF2 to be anchored more deeply into the IMM,and because AIF1 and AIF2 can form monomers or dimers(including heterodimers of AIF1/AIF2), AIF2 might have the ability to “kidnap” AIF1 by forming heterodimers, thereby affecting its capacity to translocate from the mitochondria to the cytosol, as well as its oxidoreductase activity. Under apoptotic conditions, there is usually a decrease in the mitochondrial levels of NADH. The fact that these transitional forms (monomers or dimers) are related to the oxidative stress might explain an evolutionary way to regulate the dual function of AIF during the first embryonic stages in the brain.All of this would indicate that the apoptotic function of AIF can be modulated by changes in NADH levels (Churbanova and Sevrioukova, 2008; Sevrioukova, 2009, 2011; Shetty et al., 2014). AIF is a free radical scavenger, and the de ficiency of AIF in Hq mouse brain resulted in hydroperoxides in the brain and thus aggravates oxidative stress (Klein et al., 2002; Zhu et al., 2007b).Aif2KO mice showed higher levels of markers of oxidative stress (Rodriguez et al., 2018). This suggests that local redox equilibria are affected by the changes in the AIF1/AIF2 ratio, which are caused either by the lack of AIF2 or overabundance of AIF1. However, the role of the AIF2 isoform in redox reactions is still unknown.Aifm2KO mice showed greater severity of brain damage as measured by the infarction area 72 hours after HI, while translocation to the nuclei was similar inAifm2KO mice compared to WT mice.Therefore, the most plausible explanation for the increase in the injury severity would be the increased oxidative stress in the KO mice, which might be caused by the difference in the AIF1/AIF2 ratio. Although requiring further study, AIF2 might be a better free radical scavenger than AIF1, and AIF1 and AIF2 differ in their roles in the neonatal brain after HI.(Bona et al., 1998; Fan et al., 2013). The sheep asphyxia model found that males showed an increased rate of terminal hypotension, which was not seen in females (Bennet et al.,2007). Clinical study has shown the sex-related differences of erythropoietin on neonatal HIE, with more pronounced protection in the females (Zhu et al., 2009a). All of the above sex differences indicate a “male disadvantage”; thus, the sex should be considered for the therapeutic selection. AIF could be a target for the prevention of brain injury, since AIF plays a more prominent role in neuronal cell death in males.
Sex Differences
Sex differences in perinatal brain damage have been previously reported in different studies (Nijboer et al., 2007;Zhu et al., 2013; Netto et al., 2017; Rodríguez-Fanjul et al., 2017; Charriaut-Marlangue et al., 2018). Based on the analysis of the cell death mechanisms, it has been shown that, AIF, the key to the caspase-independent pathway of cell death is more predominant in males and, in contrast, the caspase-dependent pathway is more prevalent in females(Zhu et al., 2006; Hangen et al., 2015). Bothin vitroandin vivoexperiments showed that AIF-related sex differences are not because of total AIF protein expression difference, rather AIF release from the mitochondria and translocate to the nucleus after insults. There is somewhat more pronounced AIF release in males than in females (Du et al., 2004; Zhu et al., 2006). As mentioned before, PARP-1 is a nuclear enzyme involved in DNA repair, but it is also involved in a uniquePARP-1-dependent cell death program when the DNA damage is severe (Hong et al., 2004). It is closely linked to AIF during the apoptotic process after acute neurological insults (Yu et al., 2002). Previous study has shown that the KOParp-1provided signi ficant protection overall in the group of mice. However, analysis by sex revealed that males were strongly protected in contrast to females, in which there was no significant effect. This indicates that the AIF-PARP1 pathway plays an important role in mediating neuronal cell death and sex-related differences in brain injury (Hagberg et al., 2004). Sex differences have also been reported for therapeutic hypothermia (TH), resulting in more effective long-term protection in female than in male 7-day-old rats
Potential Therapeutic Targets
Nowadays, the most common treatment for infants with HIE is based on TH applied within the first 6-10 hours after HI(Tagin et al., 2012; Douglas-Escobar and Weiss, 2015). This treatment has shown that HI brain injury can be mitigated by reducing the risk to approximately 50% (Edwards et al., 2010).However, TH is not a suitable treatment for preterm infants(Barrett et al., 2007), and it has also failed as a therapeutic strategy for severely affected children (Azzopardi et al., 2009).Furthermore, TH does not provide full neuroprotection, and about 40-50% of neonates with moderate to severe HIE still suffer severe neurological complications or die after TH(Edwards et al., 2010).
One option suggested to improve the outcome of HIE patients is to use TH together with other pharmacological adjuvants to increase neuroprotection and/or to promote neural regeneration. In this respect, low doses of recombinant human erythropoietin have shown positive effects in the longterm outcomes for infants with moderate (but not severe) HIE(Juul, 2013; Traudt et al., 2013), with only minor side effects observed (Zhu et al., 2009b). Erythropoietin has been shown to have anti-inflammatory, anti-excitotoxic, anti-oxidative,and anti-apoptotic properties (Villa et al., 2003; Wang et al.,2004). Apart from TH and erythropoietin, other substances have been proposed for improving HIE outcomes (Dixon et al., 2015), including melatonin, which has anti-apoptotic,anti-in flammatory, and anti-oxidative properties (Welin et al.,2007; Alonso-Alconada et al., 2013). Xenon, allopurinol, and magnesium sulfate have also been found to be promising for HIE treatment, but further large sample clinical studies are needed (Cilio and Ferriero, 2010; Y?ld?z et al., 2017).
Since the discovery that CypA participates in the nuclear translocation of AIF (Zhu et al., 2007a), it has been suggested to be a target for inhibiting AIF translocation after HI. A recent study showed that the AIF/CypA complex presents a good target for generating pharmacological inhibitors that block the cell death process (Thornton et al., 2017). Before that, some studies showed alternatives for that aim, such as targeting the anti-apoptotic BCL-2 family proteins (Yin et al.,2006) or preventing the translocation of AIF to the nucleus by overexpressing HSP70 (Matsumori et al., 2005). Cyclophilins influence the conformation of proteins in cells, among other functions (Kumari et al., 2013). CypA is an abundant,ubiquitously expressed protein, and it is particularly highly expressed in neurons (G?ldner and Patrick, 1996). In healthy neurons, CypA is localized both in the cytoplasm and nuclei,and it is not essential for mammalian cell viability, as indicated by the fact that CypA KO embryonic stem cells grow normally and differentiate into hematopoietic precursor cellsin vitro(Colgan et al., 2000). It has been shown that a CypA KO study reduced the total infarct volume by 46.7% and total tissue loss by 33.8% compared to the WT littermates after HI, without affecting the mitochondrial release of cell death effectors (Zhu et al., 2007a). A peptide based on the AIF and CypA sequences that corresponds to the surfaces involved in binding has already been designed (AIF370-394). This molecule acts as a cell-penetrating peptide by using the trans-activator of transcription sequence (GRKKRRQRRRPQ), which has been a very successful way to overcome the lipophilic barrier of the cellular membranes and to deliver molecules inside the cell(Vives et al., 1997; Brooks et al., 2005).In vitroexperiments in a model of glutamate-induced oxidative stress in cultured HT-22 cells have already demonstrated the efficacy and protective effect of the peptide, by acting as a competitive inhibitor of the AIF/CypA complex formation, and therefore blocking its subsequent translocation to the nucleus (Doti et al., 2014).The peptide AIF370-394 exerted pronounced neuroprotective effects while preserving its mitochondrial integrity. The effect of this peptide in a mouse HI model confirms the AIF/CypA complex as an excellent therapeutic target for preventing perinatal brain injury (unpublished data).
AIF1 has shown to be important not only for neonatal HIE but also for other organs, as the expression of AIF1 is low in the brain compared to other organs (Hangen et al.,2010b). AIF release from the mitochondria was observed in cardiomyocytes after heart failure and ischemia/reperfusion injury (Siu et al., 2007). Interestingly, muscle-specific AIF KO mice developed skeletal muscle atrophy and cardiomyopathy secondary to complex I de ficiency (Joza et al., 2005; Bano and Prehn, 2018). Therefore, therapeutic strategies targeting AIF could potentially go beyond neuroprotection.
Conclusions
The mechanisms of perinatal brain injury are not fully understood, but AIF plays a prominent role, not only due to its central function in the maintenance of mitochondrial function and oxidative levels but also because it is the key protein involved in cell death in a caspase-independent manner. AIF has already been shown to make important contributions to neuronal cell death upon HI in the immature brain. A better understanding of the mechanisms behind these processes,including the role of all AIF isoforms, and their interactions with other proteins, are needed for the development of improved and more effective treatments during the early stages of perinatal brain injury.
Author contributions:JR, TL, YX and YS wrote the original draft. CZ conceptualized, designed, reviewed and edited the review. All authors approved the final manuscript.
Con flicts of interest:The authors declare no con flicts of interest.
Financial support:This work was supported by the Swedish Research Council (2018-02667), the National Natural Science Foundation of China(31761133015, U1704281, 81901335), the Swedish Childhood Cancer Foundation (PR2018-0082), Swedish Governmental Grants to Scientists Working in Health Care (ALFGBG-717791), the Swedish Brain Foundation(FO2018-0034 and the Chinese Scholarship Council to TL (201707040025)and to YX (201507040082).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:N. Scott Litofsky, University of Missouri School of Medicine, USA.

- 中國神經(jīng)再生研究(英文版)的其它文章
- Dysfunctional glia: contributors to neurodegenerative disorders
- In flammation/bioenergetics-associated neurodegenerative pathologies and concomitant diseases: a role of mitochondria targeted catalase and xanthophylls
- Potential therapeutic effects of polyphenols in Parkinson’s disease: in vivo and in vitro pre-clinical studies
- Possible implications of dysregulated nicotinic acetylcholine receptor diffusion and nanocluster formation in myasthenia gravis
- Hydrogel-based local drug delivery strategies for spinal cord repair
- Altered physiology of gastrointestinal vagal afferents following neurotrauma