
Sensitive and rapid determination of amantadine without derivatization in human plasma by LC–MS/MS for a bioequivalence study
2018-06-20 05:50:58AhysinghBhdoriyShivprkshRthnmBhveshDsndiDhrmeshPrmrMllikSnylPrnvShrivstv
Ahysingh Bhdoriy,Shivprksh Rthnm,Bhvesh Dsndi,Dhrmesh Prmr,Mllik Snyl,Prnv S.Shrivstv
aBioanalytical Department,Synchron Research Services Pvt.Ltd,5th Floor,The Chambers,Sarkhej-Gandhinagar Highway,Bodakdev,Ahmedabad 380054,India
bDepartment of Chemistry,St.Xavier's College,Navrangpura,Ahmedabad 380009,India
cDepartment of Chemistry,School of Sciences,Gujarat University,Ahmedabad 380009,India
1.Introduction
Amantadine(AMD)is an antiviral drug having a tricyclic aliphatic ring system with a primary amino group.It is clinically used in the treatment of influenza A,Parkinsonism,hepatitis C,multiple sclerosis and drug-induced extrapyramidal reactions[1,2].The exact mechanism of action in the central nervous system is not clearly understood,but evidence suggests that AMD enhances release and reuptake balance of dopamine through antagonism of the N-methyl-D-aspartate receptor[2].This helps to reduce the symptoms of Parkinsonism[3]and multiple sclerosis[4].AMD has been extensively used in the poultry industry,particularly in chicken farming due to its antiviral properties for the treatment of influenza[5].
After oral administration,AMD gets rapidly absorbed from the gastrointestinal tract and is excreted unchanged up to 90%of the dose in the urine[6].A sizeable amount of AMD is bound to red blood cells and about 67%to plasma proteins.The peak plasma concentration of AMD reaches in 1–4 h after ingestion.It has a large apparent volume of distribution(about 5–10L/kg),signifying extensive tissue binding.AMD is metabolized to a small extent,mainly by N-acetylation,and has an elimination half-life of about 15 h[7].
Literature of the last two decades reveals several analytical methods for the determination of AMD in animal tissues[8–14],biological fluids like plasma[15–23]and urine[22,23]and also in processed food samples[14].These methods have utilized different analytical techniques like spectrophotometry[19],capillary electrophoresis[15],immunochromatography[13],micellar electrokinetic chromatography[21],ion mobility spectrometry[22],gas chromatography with flame ionization detector[23],high performance liquid chromatography(HPLC)with ultraviolet[18]and fluorescence detection[16],liquid chromatography-tandemmass spectrometry(LC–MS/MS)[14,17,20],ultra performance liquid chromatography-tandem mass spectrometry(UPLC–MS/MS)[9–12]and ultra high performance liquid chromatography coupled to high resolution LTQ orbitrap mass spectrometry[8].However,as AMD does not possess any chromophoric group,a majority of these methods require prior derivatization for sample preparation which is tedious,cumbersome and time consuming.Only three methods have analyzed AMD without derivatization in human plasma with a sensitivity of 3.9 ng/mL[17]and 20 ng/mL[20,22]respectively.Moreover,the chromatographic analysis time was≥3.5 min in these methods.A detailed summary of chromatographic methods developed for analysis of AMD in plasma samples is given in Table 1.As low concentration of AMD is expected in plasma,it is essential to develop highly sensitive and selective bioanalytical methods especially for pharmacokinetic applications.The developed method is characterized by high sensitivity,selectivity and short analysis time.The method consists of a straightforward solid phase extraction(SPE)procedure without derivatization using 200 μL of plasma samples.The chromatographic turnaround time is only 2.5 min and thus can be useful for routine sample analysis where a large number of samples need to be analyzed in a short time.The method was successfully applied for a bioequivalence study in healthy Indian subjects and the reproducibility in the measurement of subject samples was demonstrated through incurred sample reanalysis(ISR).
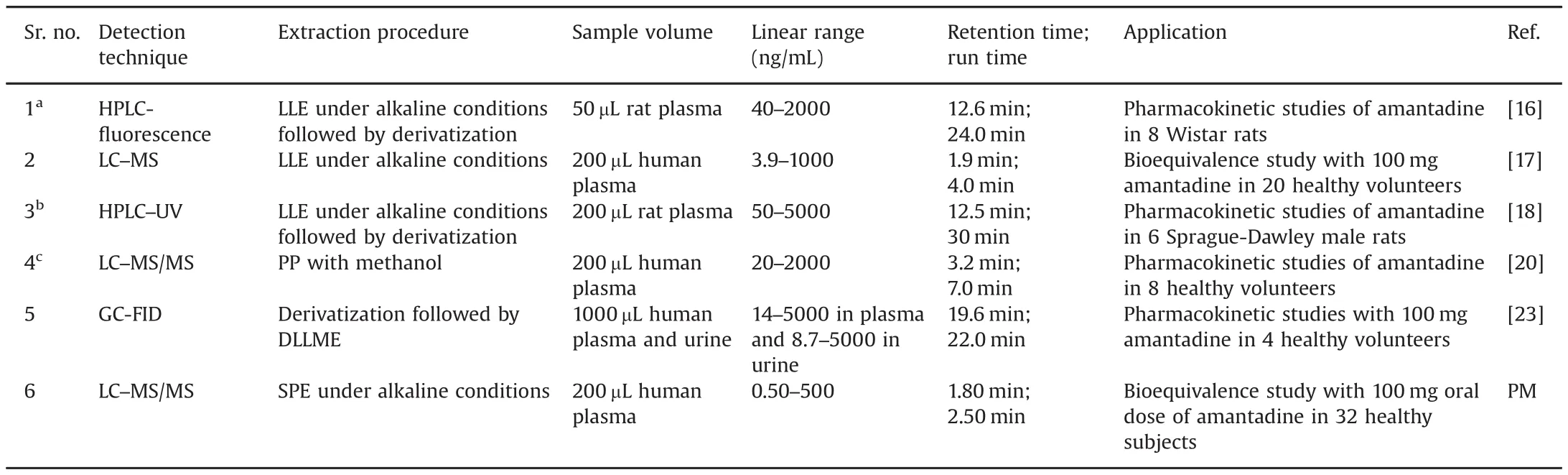
Table 1 Comparative assessment of chromatographic methods developed for analysis of amantadine in plasma and urine(1997–2017).
2.Experimental
2.1.Chemicals and materials
Reference standards of AMD(purity 99.59%)and AMD-d6(purity 99.99%)used as internal standard(IS)were obtained from Vivan Life Sciences(P)Ltd.(Mumbai,India).HPLC grade methanol,and analytical reagent grade ammonium formate,formic acid and sodium hydroxide(NaOH)were obtained from Merck Specialties Pvt.Ltd.(Mumbai,India).Strata-X-C 33μ(30mg,1mL)reversed-phase extraction cartridges were obtained from Phenomenex India(Hyderabad,India).Water used in the entire analysis was prepared using Milli-Q water purification system from Millipore(Bangalore,India).Blank human plasma in K3EDTA was obtained from Supratech Micropath(Ahmedabad,India)and was stored at-70°C until use.
2.2.Instruments and conditions
A Shimadzu Nexera X2 UHPLC equipped with Shimadzu LCMS-8040 triple quadrupole mass spectrometer(MS)detector(Shimadzu Corporation,Kyoto,Japan)was used.AMD and AMD-d6 were analyzed on Phenomenex Synergi? Hydro-RP C18(150 mm × 4.6 mm,4 μm)analytical column using an isocratic mobile phase consisting of acetonitrile and 10mM ammonium formate,pH 3.0 adjusted with 0.1%formic acid(80∶20,v/v)and delivered at a flow rate of 0.8 mL/min.The column oven temperature and autosampler temperature were maintained at 40°C and 5 °C,respectively.The injection volume was kept at 10 μL.An electrospray ionization(ESI)source operating in the positive ionization mode was used for multiple reaction monitoring(MRM)LC–MS/MS analysis.The MS conditions optimized for quantification of AMD are summarized in Table S1.Data processing was done using Shimadzu Lab Solution software.
2.3.Preparation of stock solutions,calibration standards and quality control samples
A stock solution of AMD(1000 μg/mL)was prepared by dissolving requisite amount in methanol.Working solutions were prepared by diluting the stock solution with methanol.The stock and working solutions were stored at 2–8°C.Stock solution(100 μg/mL)of AMD-d6 was prepared by dissolving 1.0 mg in 10.0 mL of methanol.Its working solution(100 ng/mL)was prepared by appropriate dilution of the stock solution in methanol.Calibration standards(CSs)and quality control(QC)samples were prepared by spiking blank plasma with working solutions.The concentration of CSs in the range of 0.50–500ng/mL and QC samplesprepared at five levels(0.05,1.50,30.0,200 and 400 ng/mL)are given in Table 2.All the samples prepared in plasma were kept at-70°C until use.
2.4.Sample extraction procedure
To an aliquot of 200 μL of spiked plasma/subject samples,50 μL of AMD-d6 working solution was added and vortexed for 10 s.The solutions were made alkaline by adding 100 μL of 0.1M NaOH and briefly vortexed.Samples were then loaded on Strata-X-C 33μ extraction cartridges which were conditioned with 1.0 mL methanol,followed by 1.0 mL water.Washing of samples was donewith 2×1.0 mL water,followed by drying of cartridges for 2.0 min by applying nitrogen(1.72×105Pa)at 2.4 L/min flow rate.The elution of AMD and AMD-d6 was done using 500μL of mobile phase solution into pre-labeled vials,and 10μL was used for injection in the chromatographic system.
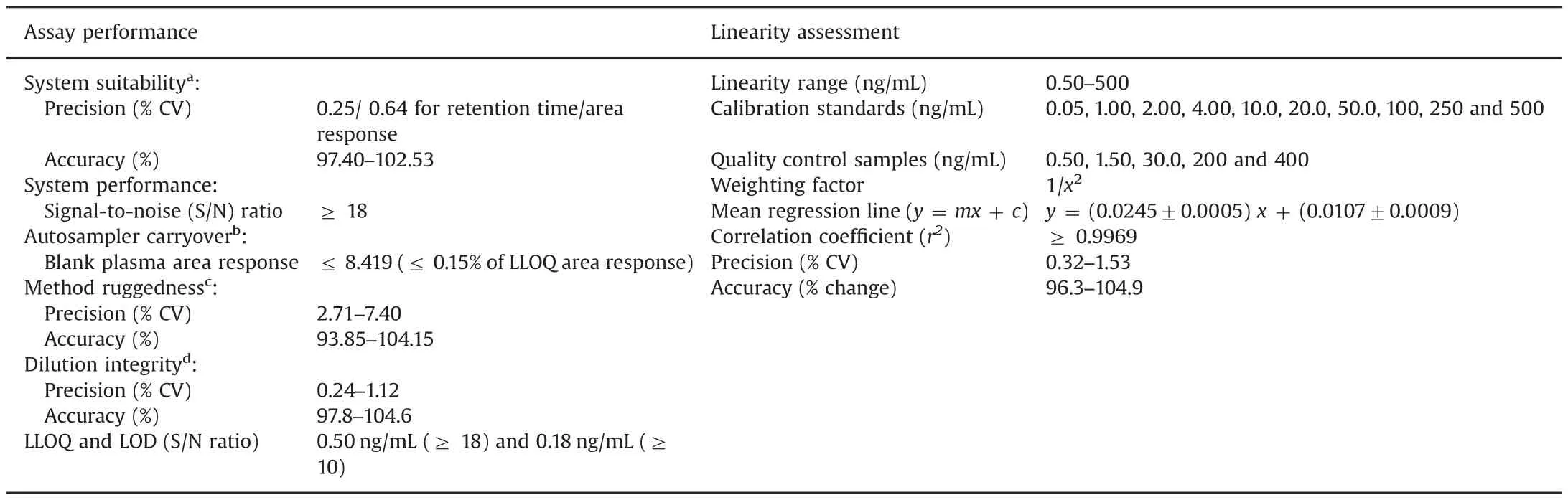
Table 2 Method performance and linearity parameters for amantadine.
2.5.Validation procedures
The method validation was performed as per the USFDA guidelines[24]and was similar to our previous work[25].The detailed procedures and their acceptance criteria are summarized in Supplementary material.
2.6.Pharmacokinetic application and method reproducibility
The developed method was used to analyze AMD plasma concentration after oral administration of single dose of a test(100 mg of amantadine hydrochloride capsules from an Indian Pharmaceutical Company)and a reference(100mg of amantadine hydrochloride capsules from Sandoz Pharmaceuticals Inc.,Princeton,NJ,USA)formulation to 32 healthy Indian subjects under fasting.The study was conducted as per International Conference on Harmonization,E6 Good Clinical Practice Guidelines[26].The experimental details for the study are given in Supplementary material.The pharmacokinetic parameters of AMD were estimated by non-compartmental analysis using WinNonlin?software version 5.3(Certara,Princeton,NJ 08540,USA).Method reproducibility was ascertained through ISR using 134 subject samples having concentration near the Cmaxand the elimination phase in the pharmacokinetic pro file of the drug and its metabolites.According to the acceptance criterion,at least two-thirds of the original and repeat results should be within 20%of each other[27].
3.Results and discussion
3.1.LC–MS/MS method development
Till date there are only two chromatographic methods based on mass spectrometric detection to analyze AMD in human plasma without prior derivatization[17,20].Feng et al.[20]determined AMD together with some common medications like paracetamol,caffeine and chlorpheniramine maleate using protein precipitation(PP)for a pharmacokinetic study.However,the recovery was very low(~52%)and the sensitivity of the method was 20 ng/mL for AMD.A much improved LC–MS procedure was reported by Wang and co-workers[17]with a linear concentration range of 3.9–4000 ng/mL using liquid-liquid extraction(LLE).The chromatographic analysis time was 4.0 min under isocratic elution,while both the methods used a general internal standard for area ratio measurements.Thus,based on the outcome of these reports we developed a highly sensitive,selective,rapid and robust method using UHPLC–MS/MS instrumentation and SPE for sample processing employing a deuterated IS,which can give a good measure of control for extraction and ionization variability.
As both AMD and AMD-d6 have a primary amino group,mass spectrometry was performed in the positive ionization mode using ESI.Under the optimized mass spectrometric conditions,intense protonated molecular ions[M+H]+were obtained atm/z152.1 and 158.0 for AMD and AMD-d6,respectively in the full-scan mode(Q1).The product ion spectrum(Q3)provided highest signals atm/z135.1 and 141.1 for AMD and AMD-d6,respectively(Fig.S1).These stable product ions were obtained by the elimination of amino groups from their precursor ions.In the present work,sample cleanup was initiated on two SPE cartridges,namely,Strata-X-C and Oasis HLB,for quantitative and precise extraction recovery with minimal matrix interference.Initial trials with PP using acetonitrile and methanol yielded poor recovery of AMD with considerable matrix interference(42%–59%),while LLE with ethylacetate,n-hexane,methyltert-butylether and dichloromethane alone and in combination afforded somewhat higher recovery(~72%)but was inconsistent at lower concentrations(0.05 and 1.50 ng/mL).With SPE under neutral conditions it was difficult to completely retain AMD(pKa 9.0)[10]and AMD-d6 during the washing step on both the cartridges.Although the recovery obtained was precise at all QC levels,there was a loss of about 15%–18%.Thus,the solutions were made alkaline prior to loading,which led to considerable improvement in recovery(>87%).Both the cartridges provided quantitative recovery,but Strata-X-C specifically used for weakly basic compounds(pKa 8–10)gave higher recoveries compared to Oasis HLB,and hence was used in the present work.
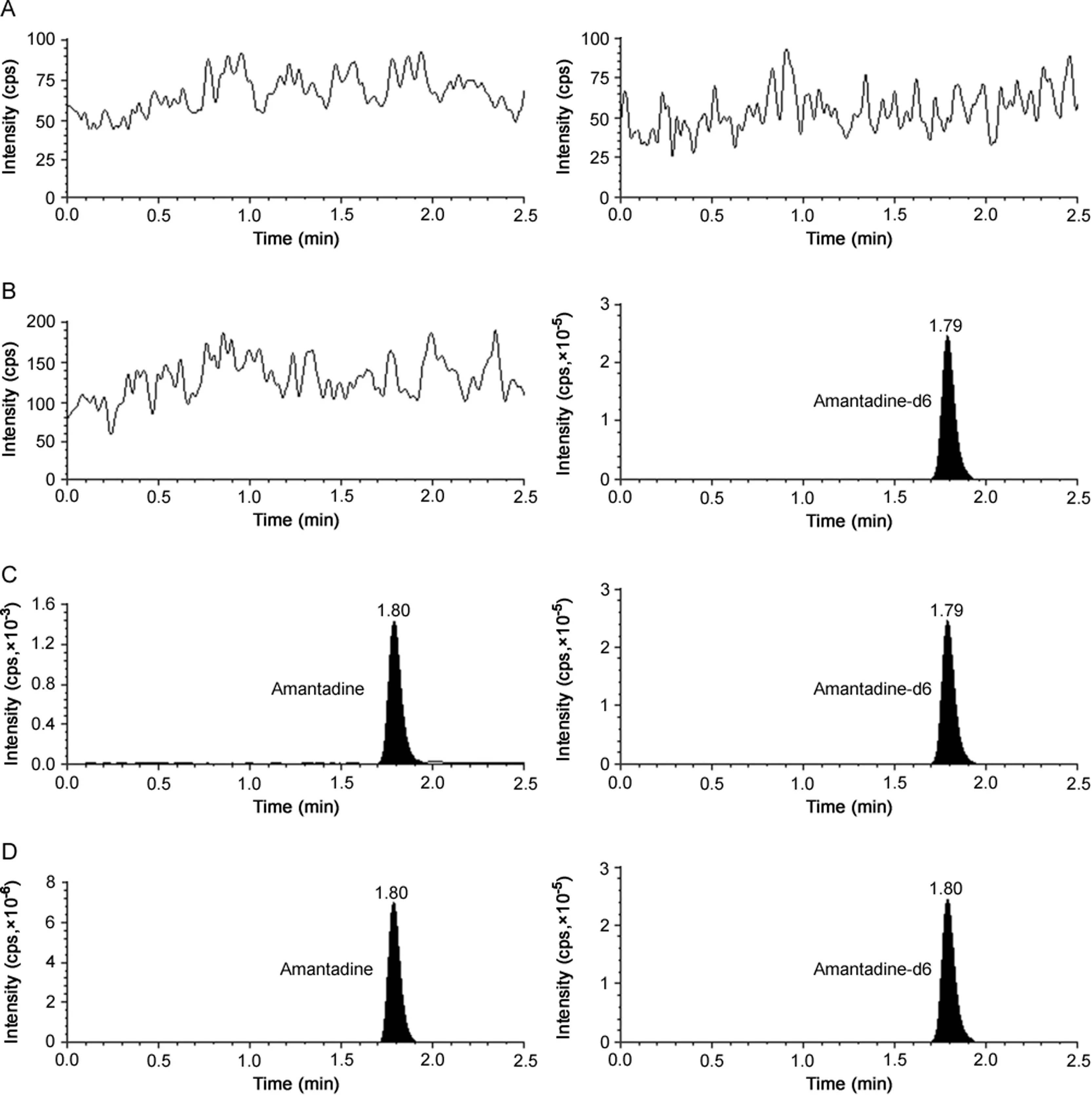
Fig.1.Representative chromatograms of(A)double blank plasma(without amantadine and amantadine-d6),(B)blank plasma spiked with amantadine-d6(100 ng/mL),(C)amantadine(0.50 ng/mL)and amantadine-d6(100 ng/mL)and(D)real subject sample at Cmaxafter oral administration of 100 mg dose of amantadine.
Different reversed-phase columns were assessed for a reliable and reproducible analysis of AMD.Columns tested included Kromasil C18(150 mm × 4.6mm,3.5 μm),Hypurity C18(100mm ×4.6 mm,5μm),Zorbax Eclipse XDB C18(150 mm × 4.6 mm,5μm)and Synergi Hydro-RP C18(150 mm × 4.6 mm,4 μm).Various mobile phase combinations(50∶50,60∶40,70∶30 and 80∶20,v/v)of acetonitrile/methanol and 5–20 mM ammonium formate at different pH(2.5–6.5)were tried during development stage.Better results were obtained with lower pH values,which correlated with the capacity factor(K).With increase in pH values(4.5–6.5),theKvalues were in the range of 0.4–0.6,possibly due to the formation of unionized species which had little retention.A similar trend was observed when the concentration of ammonium formate was increased from 5.0 to 20 mM.Further the impact of mobile composition showed considerable reduction in analyte response when the aqueous proportion was greater than 30%.Acetonitrile was selected ahead of methanol as it provided much better peak shape.Under the optimized mobile phase conditions of acetonitrile and 10 mM ammonium formate(pH 3.0),AMD was not adequately retained on Kromasil C18and Hypurity C18columns,while the response and peak shape were not acceptable on Zorbax Eclipse XDB C18.As a result,Synergi Hydro-RP C18column which provided adequate retention,sufficient response and good peak shape was employed for further study.Additionally,use of deuterated IS helped to ensure acceptable method performance based on similar extraction recovery,chromatographic retention time and ionization response in ESI-MS/MS.The retention time for AMD and AMD-d6 was 1.80 and 1.79 min,respectively,in a total run time of 2.5 min(Fig.1).The reinjection reproducibility(%CV)in the measurement of retention time was≤1.2%.
The developed method was more sensitive by about 8[17]and 40[20]times compared to existing LC–MS methods in humanplasma.Moreover,the analysis time was 1.5 times less than the method reported by Wang et al.[17],which can be of advantage when large numbers of samples are to be analyzed,especially in a clinical setting.A comparative assessment of all chromatographic methods developed in the last two decades in plasma and urine is presented in Table 1.
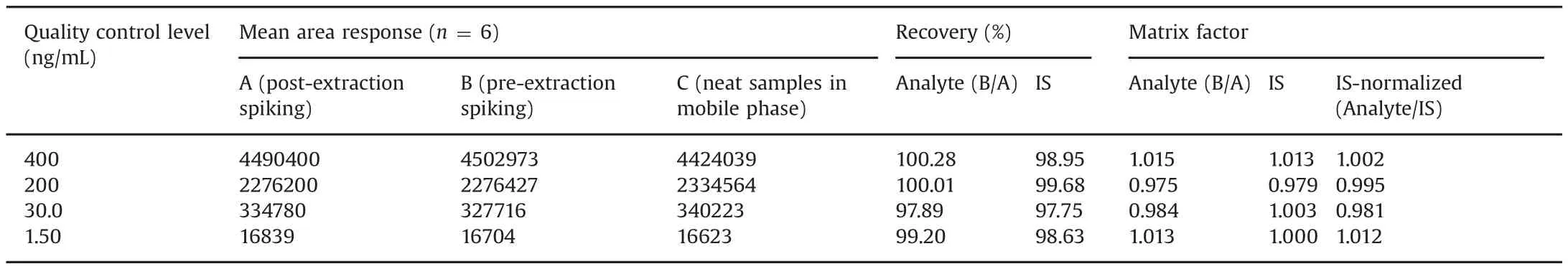
Table 3 Extraction recovery and matrix factor for amantadine from human plasma.
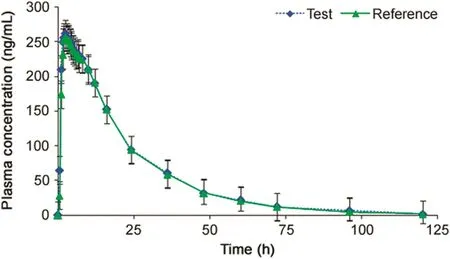
Fig.2.Mean plasma concentration-time pro file of amantadine after oral administration of 100 mg(test and reference)capsule formulation to 32 healthy Indian subjects.
3.2.Assay validation results
The results for system suitability,autosampler and column carryover,ruggedness and dilution integrity suggest acceptable assay performance as evident from the data presented in Table 2.The selectivity of the method is evident from the chromatograms of double blank plasma,plasma spiked with AMD-d6,AMD at 0.50 ng/mL concentration and real subject sample at Cmaxin Fig.1.No interference due to endogenous components was observed at the retention time of AMD and AMD-d6.Furthermore,none of the commonly used medications by human volunteers interfered at their retention times.The calibration curves showed good linearity over the established concentration range of 0.50–500 ng/mL(r2≥ 0.9969)for AMD.The mean values for slope,intercept,accuracy and precision data in the measurement of calibrator concentrations are shown in Table 2.The intra-batch precision(%CV)ranged from 0.56% to 5.42% and the accuracy was within 98.47%–105.72%for AMD.Similarly for inter-batch experiments,the precision varied from 1.27%to 4.23%and the accuracy was within 98.86%–105.21%(Table S2).
The mean extraction recovery and IS-normalized matrix factors(MFs)for AMD are presented in Table 3.Highly precise extraction recovery in the range of 97.89%–100.28%was obtained across QC levels.The mean recovery of AMD-d6 was 98.75%.As presence of unmonitored,co-eluting compounds from the matrix can directly impactthe overallperformance ofa validated method,itis necessary to evaluate MFs to assess the matrix effect.The IS-normalized MFs ranged from 0.981 to 1.012.Matrix effect was also checked in lipemic and haemolysed plasma samples together with normal K3EDTA plasma.This was determined by examining the precision(%CV)values of the slopes of the calibrations curves prepared from eight different plasma lots,which included six K3EDTA,one lipemic and one haemolysed plasma samples.The%CV of the slopes of calibration lines for relative matrix effect in eight different plasma lots was 1.52%,which is within the acceptance criteria of 3%–4%.
Stock solutions kept for short-term and long-term stability as well as spiked plasma solutions showed no evidence of degradation under all studied conditions.Samples for short-term stability remained stable up to 8h,while the stock solutions of AMD and AMD-d6 were stable for minimum of 18 days at refrigerated temperature of 5°C.As a substantial amount of AMD is bound to red blood cells,whole blood stability of AMD was also evaluated by spikingbloodsamples(500μL)withAMDat 0.15 and 400ng/mL concentrations for 2.0 h.The detailed results for stability studies are presented in Table S3.
3.3.Application to a bioequivalence study and ISR results
To the best of our knowledge there have been no reports on the pharmacokinetics of AMD in Indian subjects.Thus,the developedmethod was applied to determine plasma AMD concentration in 32 healthy Indian subjects after oral administration of 100 mg AMD capsules under fasting.The mean plasma concentration–time pro files obtained for the test and reference formulations are shown in Fig.2.Table 4 summarizes mean values of pharmacokinetic parameters for both the formulations.Comparison of the results obtained with a similar study using 100 mg dose of AMD in 20 Chinese subjects[17]showed no significant change inTmaxandt1/2values.However,theCmaxvalues obtained in the present study were lower,while AUC values were somewhat higher than their results.Nevertheless,the ratios of mean log-transformed parameters,Cmax,AUC0–120hand AUC0-infand their 90%confidence intervals ranged from 101.23%to 102.63%and 96.14%to 108.42%for AMD,respectively,which is within the acceptance criterion of 80%–125%.These results confirm the bioequivalence of the test formulation with the reference product in terms of rate and extent of absorption.Furthermore,the assay reproducibility test performed with 134 incurred samples showed%change within±12%of the initial results,which confirms the reproducibility of the newly developed method.

Table 4 Mean pharmacokinetic(±SD)parameters,comparison of treatment ratios and 90%CIs of natural log(Ln)-transformed parameters following oral administration of 100 mg of amantadine tablet formulation in 32 healthy Indian subjects under fasting.
4.Conclusions
The proposed LC–MS/MS assay for the quantitation of AMD in human plasma was developed and fully validated as per current regulatory guidelines.This method can be useful for the analysis of large numbers of samples as it uses a simple extraction procedure without derivatization,requires low sample volume,is highly selective,and has a short assay time.Further,the method shows excellent accuracy and precision,reproducible recoveries and minimal matrix effects.In addition,the method was successfully applied to determine plasma AMD concentration in a bioequivalence study with healthy Indian subjects for the first time.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgments
The authors wish to thank the management of Synchron Research Services Pvt.Ltd,Ahmedabad to carrying out this research.
Appendix A.Supplementary material
Supplementary data associated with this article can be found in the online version at doi∶10.1016/j.jpha.2017.10.003.
[1]I.T.Prudhomme,O.Zoueva,J.M.Weber,Amantadine susceptibility in influenza A virus isolates∶determination methods and lack of resistance in a Canadian sample,1991–94,Clin.Diagn.Virol.8(1997)41–51.
[2]W.Xu,N.Wei,Y.Xu,et al.,Does amantadine induce acute psychosis?A case report and literature review,Neuropsychiatr.Dis.Treat.12(2016)781–783.
[3]J.Romrell,H.H.Fernandez,M.S.Okun,Rationale for current therapies in Parkinson's disease,Expert Opin.Pharmacother.4(2003)1747–1761.
[4]L.B.Krupp,Fatigue in multiple sclerosis∶definition,pathophysiology and treatment,CNS Drugs 17(2003)225–234.
[5]D.Cyranoski,China's chicken farmers under fire for antiviral abuse,Nature 435(2005)(1009-1009).
[6]J.M.Meythaler,R.C.Brunner,A.Johnson,et al.,Amantadine to improve neurorecovery in traumatic brain injury-associated diffuse axonal injury∶a pilot double-blind randomized trial,J.Head Trauma Rehabil.17(2002)300–313.
[7]SYMMETREL?(Amantadine Hydrochloride),Prescribing Information,NOVARTIS Pharm aceuticals Australia Pty Limited,54 Waterloo Road,North Ryde,NSW,Australia,2014.〈http∶//www.guildlink.com.au/gc/ws/nv/pi.cfm?pro duct=nvpsymor11014〉.(Accessed March 2017).
[8]H.Yan,X.Liu,F.Cui,et al.,Determination of amantadine and rimantadine in chicken muscle by QuEChERS pretreatment method and UHPLC coupled with LTQ Orbitrap mass spectrometry,J.Chromatogr.B 938(2013)8–13.
[9]Y.L.Wu,R.X.Chen,Y.Xue,et al.,Simultaneous determination of amantadine,rimantadine and memantine in chicken muscle using multi-walled carbon nanotubes as a reversed-dispersive solid phase extraction sorbent,J.Chromatogr.B 965(2014)197–205.
[10]Z.C.Liu,F.Yang,M.Yao,et al.,Simultaneous determination of antiviral drugs in chicken tissues by ultra high performance liquid chromatography with tandem mass spectrometry,J.Sep.Sci.38(2015)1784–1793.
[11]P.Mu,N.Xu,T.Chai,et al.,Simultaneous determination of 14 antiviral drugs and relevant metabolites in chicken muscle by UPLC–MS/MS after QuEChERS preparation,J.Chromatogr.B 1023–1024(2016)17–23.
[12]Q.Zhang,C.Xiao,W.Wang,et al.,Chromatography column comparison and rapid pretreatment for the simultaneous analysis of amantadine,rimantadine,acyclovir,ribavirin and moroxydine in chicken muscle by ultra high performance liquid chromatography and tandem mass spectrometry,J.Sep.Sci.39(2016)3998–4010.
[13]S.Wu,F.Zhu,L.Hu,et al.,Development of a competitive immunochromatographic assay for the sensitive detection of amantadine in chicken muscle,Food Chem.232(2017)770–776.
[14]Y.Tsuruoka,T.Nakajima,M.Kanda,et al.,Simultaneous determination of amantadine,rimantadine,and memantine in processed products,chicken tissues,and eggs by liquid chromatography with tandem mass spectrometry,J.Chromatogr.B 1044(2017)142–148.
[15]S.Nagaraj,S.V.Rahavendran,H.T.Karnes,Visible diode laser induced fluorescence detection for capillary electrophoretic analysis of amantadine in human plasma following precolumn derivatization with Cy5.29.OSu1,J.Pharm.Biomed.Anal.18(1998)411–420.
[16]Y.Higashi,I.Uemori,Y.Fujii,Simultaneous determination of amantadine and rimantadine by HPLC in rat plasma with pre-column derivatization and fluorescence detection for pharmacokinetic studies,Biomed.Chromatogr.19(2005)655–662.
[17]P.Wang,Y.Z.Liang,B.M.Chen,et al.,Quantitative determination of amantadine in human plasma by liquid chromatography–mass spectrometry and the application in a bioequivalence study,J.Pharm.Biomed.Anal.43(2007)1519–1525.
[18]C.Shuangjin,F.Fang,L.Han,et al.,New method for high-performance liquid chromatographic determination of amantadine and its analogues in rat plasma,J.Pharm.Biomed.Anal.44(2007)1100–1105.
[19]A.M.Mahmoud,N.Y.Khalil,I.A.Darwish,Selective spectrophotometric and spectro fluorometric methods for the determination of amantadine hydrochloride in capsules and plasma via derivatization with 1,2-naphthoquinone-4-sulphonate,Int.J.Anal.Chem.(2009)1–8(Article ID 810104).
[20]S.Feng,Y.Tian,Z.Zhang,et al.,Rapid simultaneous determination of paracetamol,amantadine hydrochloride,caffeine and chlorpheniramine maleate in human plasma by liquid chromatography/tandem mass spectrometry,Arzneimittelforschung 59(2009)86–95.
[21]H.H.Yeh,Y.H.Yang,S.H.Chen,Simultaneous determination of memantine and amantadine in human plasma as fluorescein derivatives by micellar electrokinetic chromatography with laser-induced fluorescence detection and its clinical application,Electrophoresis 31(2010)1903–1911.
[22]M.Saraji,T.Khayamian,S.Mirmahdieh,et al.,Analysis of amantadine in biological fluids using hollow fiber-based liquid–liquid–liquid microextraction followed by corona discharge ion mobility spectrometry,J.Chromatogr.B 879(2011)3065–3070.
[23]M.A.Farajzadeh,N.Nouri,A.A.A.Nabil,Determination of amantadine in biological fluids using simultaneous derivatization and dispersive liquid–liquid microextraction followed by gas chromatography- flame ionization detection,J.Chromatogr.B 940(2013)142–149.
[24]Guidance for Industry,Bioanalytical Method Validation,US Department of Health and Human Services,Food and Drug Administration Centre for Drug Evaluation and Research(CDER),Centre for Veterinary Medicine(CVM),May 2001.
[25]P.Sharma,P.A.Shah,M.Sanyal,et al.,Challenges in optimizing sample preparation and LC-MS/MS conditions for the analysis of carglumic acid,an N-acetyl glutamate derivative in human plasma,Drug Test.Anal.7(2015)763–772.
[26]Guidance for Industry∶ICH E6 Good Clinical Practice,U.S.Department of Health and Human Services,Food and Drug Administration,Centre for Drug Evaluation and Research(CDER),Centre for Biologics Evaluation and Research(CBER),1996.
[27]M.Yadav,P.S.Shrivastav,Incurred sample reanalysis∶a decisive tool in bioanalytical research,Bioanalysis 3(2011)1007–1024.
 Journal of Pharmaceutical Analysis2018年3期
Journal of Pharmaceutical Analysis2018年3期
- Journal of Pharmaceutical Analysis的其它文章
- JPA Prize in 2016
- Physics,chemistry,and Hirshfeld surface analyses of gamma-irradiated thalidomide to evaluate behavior under sterilization doses
- Enrichment and immobilization of macromolecular analytes on a porous membrane utilizing permeation drag
- Enhancing the dissolution of phenylbutazone using Syloid?based mesoporous silicas for oral equine applications
- Identi fication of three kinds of Plumeria flowers by DNA barcoding and HPLC specific chromatogram
- Evaluation of physicochemical properties as supporting information on quality control of raw materials and veterinary pharmaceutical formulations