
Effect of Substituents on the Optical Properties of 3(5)-(9-Anthryl)Pyrazole
2014-10-14 03:45:12WANGKunPengWANGChangSheng
物理化學(xué)學(xué)報 2014年5期
WANG Kun-Peng WANG Chang-Sheng
(School of Chemistry and Chemical Engineering,Liaoning Normal University,Dalian 116029,Liaoning Province,P.R.China)
Effect of Substituents on the Optical Properties of 3(5)-(9-Anthryl)Pyrazole
WANG Kun-Peng WANG Chang-Sheng*
(School of Chemistry and Chemical Engineering,Liaoning Normal University,Dalian 116029,Liaoning Province,P.R.China)
Abstract: The ground state(S0)structures of 3(5)-(9-anthryl)pyrazole and its derivatives were obtained using the density functional theory(DFT)B3LYP/6-31G(d)method.The first singlet excited state(S1)structures were optimized using the singlet-excitation configuration interaction(CIS)/6-31G(d)method.The absorption and emission spectra were then evaluated using the time-dependent density functional theory(TD-DFT)B3LYP method with the 6-311++G(d,p)basis set.Our calculation results reveal that for all the derivatives(electron-withdrawing groups or electron-donating groups)the calculated absorption and fluorescence emission wavelength values all show red shifts compared with the parent 3(5)-(9-anthryl)pyrazole.We also find that compared with the parent 3(5)-(9-anthryl)pyrazole,the derivatives with―R=―BH2,―CCl3,―CHO,―NH2are good candidates for longer absorption wavelength materials and for longer fluorescence emission wavelength materials.
Key Words:Absorption spectrum;Fluorescence emission spectrum;3(5)-(9-Anthryl)pyrazole;Excited state
1 Introduction
The design and synthesis of organic optical materials have attracted intensive attention because of their potential applications in organic light-emitting diodes(OLEDs).Much effort has been made on the multicolor patterning of organic luminescent molecules with ordered micro-and nano-scopic features as a result of their applications in full-color display and other related areas.1-19Mizukamiet al.1found that a helical 3,3′-di-tert-butylsalen-zinc(II)complex,[Zn2],has a red-shifted fluorescence as compared to that of[Zn],a half-structured mononuclear complex of[Zn2].Baderet al.2reported the syntheses and electrochemical properties of four oligothiophene derivativeswith the tricyanovinyl group and suggested that these materials might be suitable for n-type,and possibly for ambipolar,transport.Yamaguchi et al.3designed and synthesized a series of B,B′,B″-trianthryl-N,N′,N″-triarylborazine derivatives bearing various p-substituted phenyl groups and observed significant bundle effects in the photophysical and electrochemical properties of these compounds.Murata et al.4reported efficient molecular organic light-emitting diodes composed of novel silols derivatives as an electron transporting layer and an emissive layer.Tang et al.5prepared a series of 2,3,4,5-tetraphenylsiloles with different 1,1-substituents and observed that with an increase in the electronegativity of 1,1-substituents of the silols,the absorption and emission wavelengths of the silols bathochromically shifted.Sapochak et al.6carried out theoretical and experimental investigations on the molecular and electronic structure of the 8-hydroxyquinoline chelate of zinc(II)and related the results to OLED performance.Brinkmann et al.7investigated the structures and the correlation between intermolecular interactions and optical properties in various metaloquinolate tris(8-hydroxyquinoline)aluminum(Alq3)systems,including solution,amorphous thin films,and different crystalline forms,and showed that the length of interligand contacts between neighboring Alq3molecules as well as the molecular density of the packing plays an important role in influencing the spectral position of fluorescence.The molecular orbital study of the first excited state of the OLED material Alq3was carried out by Schlegel et al.8Based on the structure of the excited state,they predicted an emission wavelength of 538 nm,which is comparable to 514 nm observed experimentally for solution phase photoluminescence.Geng et al.9investigated the electronic structure and transport properties of a p-stacking molecular chain using the first-principles density functional theory approach combined with Green′s function method.Su et al.10carried out a DFT/TD-DFT study on the electronic structures and optoelectronic properties of several blue-emitting iridium(III)complexes and found that the properties of the ligands had great influence on the photophysical properties,such as energy gap,absorption spectra,emission spectra,etc.Zhang et al.11investigated the optical properties of the phosphorescent trinuclear copper(I)complexes of pyrazolates theoretically and found that the short intermolecular Cu…Cu distance played an important role in the emission spectra of the verticaland tilting-movement dimers.An theoretical study on symmetric and asymmetric spirosilabifluorene derivatives was also carried out and an excellent agreement with the experiment data on their optical properties was obtained.12Wang et al.13-14reported the emission properties of the polymorphs and pseudopolymorphs of N,N-di(n-butyl)quinacridone and N,N-di(n-cetyl)quinacridone and found that the crystal phases with stronger π-π interactions showed the emission maximum at a longer wavelength region while that with relatively weaker π-π interactions exhibited an emission maximum at a shorter wavelength region.Although considerable progress has been made in organic luminescent materials with different structures,it is still essential to achieve a molecular-level understanding of the relationship between the electronic structures and the resulting optical properties.
The organic molecule 3(5)-(9-anthryl)pyrazole has been used as building blocks to construct different luminescent single crystal.20-22In order to understand the optical properties of 3(5)-(9-anthryl)pyrazole,we here reported our research on the geometrical structure,the gap between the highest occupied molecular orbital(HOMO)and the lowest unoccupied molecular orbital(LUMO),and the absorption/emission spectra of 3(5)-(9-anthryl)pyrazole at the ground and excited states with the DFT and CIS methods.We displayed the relationship between the optical properties and electronic structures of the frontier molecular orbitals(FMOs),explored the influence of the substitutions on the absorption and emission wavelengths.We hope that the result obtained would be helpful for understanding connection between the fluorescence characters and electronic structures and helpful for the find of new fluorescence materials.
2 Calculation methods
ANP possesses two possible structures ANP-1 and ANP-2(Fig.1).The ground state S0structures of ANP-1 and ANP-2 were obtained by using the density functional theory23B3LYP/6-31G(d)method.B3LYP/6-311++G(d,p)//B3LYP/6-31G(d)calculation showed that ANP-1 was a little more stable than ANP-2.The singlet excited state S1structures of ANP-1 and ANP-2 were optimized by using the CIS method24with 6-31G(d)basis set.Based on the structures obtained,their absorption and emission spectra were calculated by the TD-DFT B3LYP method with 6-311++G(d,p)basis sets.In order to examine the effect of substituents on the absorption and emission spectra,several derivatives of ANP-1 were designed by replacing the hydrogen atom on the C7 of ANP-1 with several electron-withdrawing or electron-donating groups.The ground state S0structures and the singlet excited state S1structures of these derivatives were optimized by using B3LYP/6-31G(d)and CIS/6-31G(d)methods,respectively.Based on the structures obtained,the absorption and emission spectra of these derivatives were then obtained by using TD-DFT B3LYP method with 6-311++G(d,p)basis sets.The solvent affect was considered by using PCM model25in TD-DFT B3LYP calculations.
3 Results and discussion
3.1 Geometric structures and electronic structures of ANP-1 and ANP-2
The selected structure parameters of ANP-1 and ANP-2 are listed in Table 1.Compared with the ground state S0,some bond lengths in the excited state S1are lengthened,some others are shortened.The bond lengths of C1―C6,C2―C3,and C4―C5 are all shortened from the ground state S0to the excited state S1for both ANP-1 and ANP-2,while the bond lengthsof C1―C2,C5―C6,C3―C7,and C4―C11 are all lengthened.The bond lengths between the adjacent atoms except H in the pyrazole ring of ANP-1 and ANP-2 are all shortened with the exception of the bond length C12―C19 in ANP-2 which is lengthened slightly from 0.1423 to 0.1424 nm.The dihedral anglesDN15C12C11C10become smaller for both ANP-1 and ANP-2 from the ground stateS0to the excited stateS1,indicating a better planar nature of the excited stateS1.What is more,their bond lengths C11―C12 are within the range from 0.1485 to 0.1472 nm,which are longer than the standard double bond(C=C,0.134 nm)and shorter than the standard single bond(C―C,0.154 nm),indicating a certain double bond nature.Thus conclusion could be drawn that a larger conjugation system has formed fromS0toS1,which will facilitate the free movement of electron cloud and electron transfer from the pyrazole fragment to the anthracene fragment.
The electron density contours of HOMO-1,HOMO,LUMO,LUMO+1 of ANP-1 and ANP-2(Fig.2)show that HOMO is mainly distributed on the anthracene ring part with a small quantity of distribution on the pyrazole fragment,LUMO is mainly distributed on the anthracene ring part and much less distributed on the pyrazole fragment.Based on the fact that the lowest lying excited state usually corresponds to an excitation from HOMO to LUMO,we can make a good explanation of the ANP bond length variation by analyzing the FMOs.The bonds C1―C6,C2―C3,and C4―C5 are antibonding on the HOMO for both ANP-1 and ANP-2,while on the LUMO,they are bonding in these regions.Therefore,these bonds are shortened on the excited stateS1.The bonds C1―C2,C5―C6,C3―C7,and C4―C11 are antibonding on the LUMO,while they are bonding on the HOMO.These bonds hence become longer on the excited stateS1.
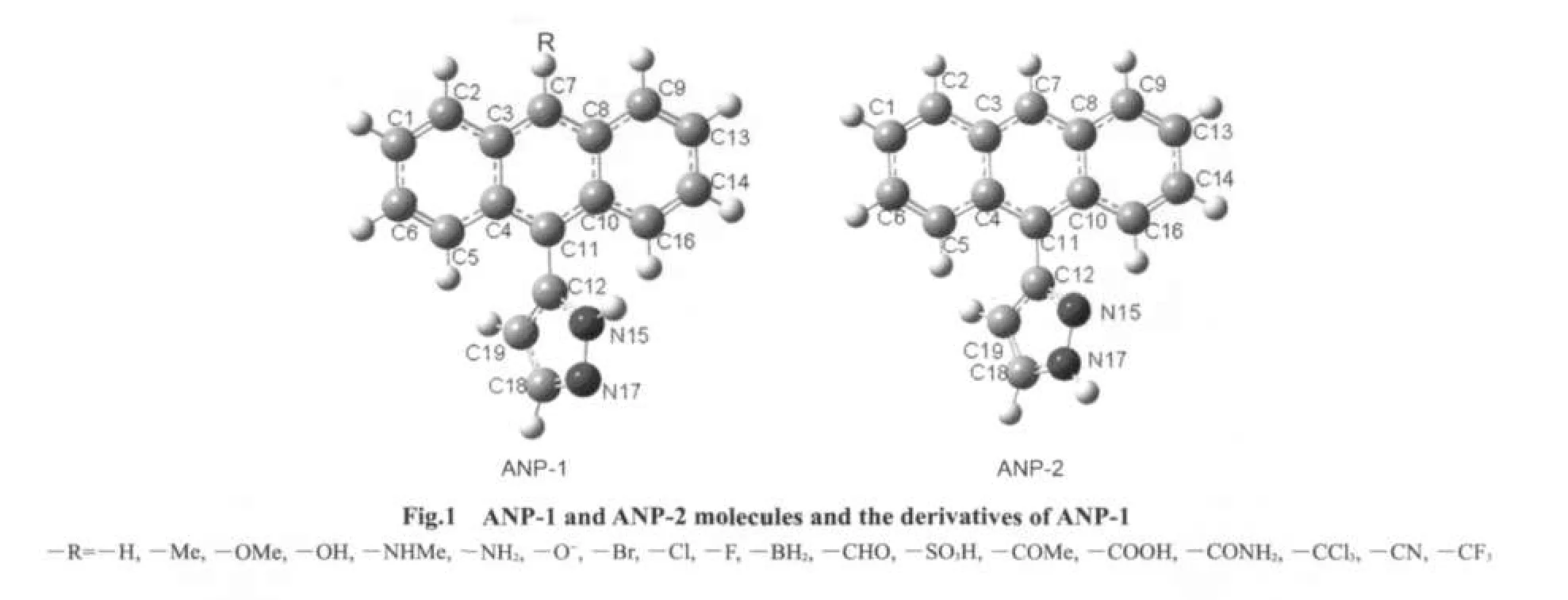
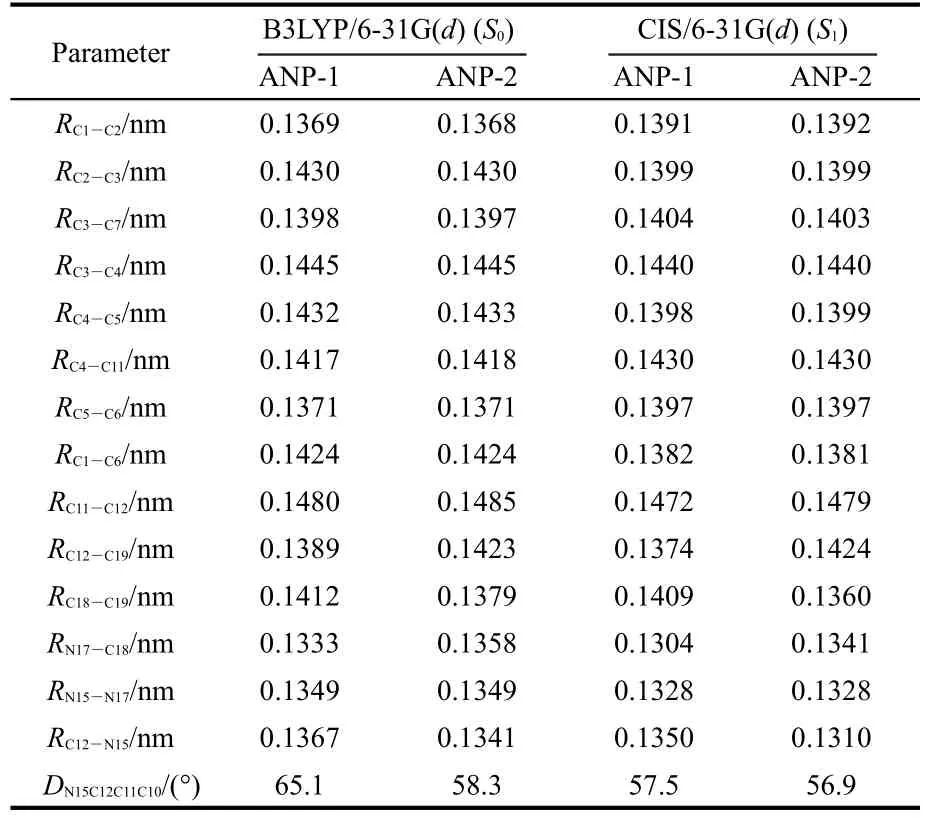
Table 1 Optimized structural parameters ofANP-1 andANP-2
3.2 Effect of substituents on the optical properties
3.2.1 The frontier molecular orbital(FMO)analysis
The HOMO and LUMO energies of ANP-1 and its derivatives are listed in Table 2.As seen from Table 2,both HOMO and LUMO energies are obviously changed by replacing the hydrogen atom on the C7 of ANP-1 with an electron-withdrawing or electron-donating group.Table 2 shows that compared with the parent molecule ANP-1(―R=―H),replacing the hydrogen atom with an electron-withdrawing group(―R=―BH2,―CHO,―SO3H,―COMe,―COOH,―CONH2,―CCl3,―CF3,―CN)lowers both the HOMO energy(EHOMO)and the LUMO energy(ELUMO),andELUMOis lowered more thanEHOMO,resulting in a smaller energy gapsEgcompared with the energy gap(3.472 eV)of ANP-1,suggesting a red-shift absorption wavelength.However,substituting the hydrogen atom with an electron-donating group(―R=―Me,―OH,―OMe,―NH2,―NHMe)raises bothEHOMOandELUMO,butEHOMOis raised more,resulting in a smaller energy gap compared with that of ANP-1,also suggesting a red-shift absorption wavelength.
3.2.2 Absorption spectra
Table 3 displays the calculated absorption wavelength(λ),the oscillator strength(f),transition assignment,and the main CI expansion coefficients of ANP-1 and its derivatives.It has been known that a large oscillator strengthfusually corresponds to a large experimental absorption coefficient or a strong fluorescence.It can be seen from Table 3 that,for all thederivatives,whatever the substituent is an electron-withdrawing group or an electron-donating group,the calculated absorption wavelength values all show a red shift as compared with ANP-1,consistent with the FMO analysis above.Table 3 shows that the derivative with―R=―NH2possesses a relatively longer absorption wavelength(442 nm)than the derivative with―R=―NHMe(428 nm),the derivative with―R=―OH possesses a relatively longer absorption wavelength(424 nm)than the derivative with―R=―OMe(413 nm),the derivative with―R=―CHO possesses a relatively longer absorption wavelength(447 nm)than the derivative with―R=―COMe(410 nm),showing that the methyl group is not a good candidate for designing an optical material possessing a longer absorption wavelength.Table 3 also shows that among the derivatives with―R=―F,―Cl,―Br,the derivative with―R=―Br exists the longest absorption wavelength(415 nm)whereas the derivative with―R=―F shows the shortest absorption wavelength(409 nm),suggesting that the electronegativity may play a role:a more electronegative group may result in a shorter absorption wavelength,or,in another word,a more electropositive group may result in a longer absorption wavelength.This conclusion is further confirmed by the fact that the derivative with―R=―CCl3has a longer absorption wavelength than the one with―R=―CF3.Among the substituents considered,we find that the derivatives with―R=―BH2,―CCl3,―CHO,and―NH2have relatively long absorption wavelength,three of them belong to the electron-withdrawing group,suggesting that if one want to design and synthesize an optical material possessing a longer wavelength,one can substitute the hydrogen atom of the parent molecule with electron-withdrawing group,especially with ―BH2,―CCl3,―CHO.
The data in the third column of Table 3 are the absorption wavelength in CHCl3solvent.The data in the fifth column of Table 3 are the oscillator strengths in CHCl3solvent.These data show that the solvent CHCl3leads to a further red shift for the absorption wavelength values with the red-shifted extent 3-13 nm except the derivative with―R=―O-.Moreover,oscillator strengthsfin CHCl3solvent are all larger than the counterparts in gas phase.
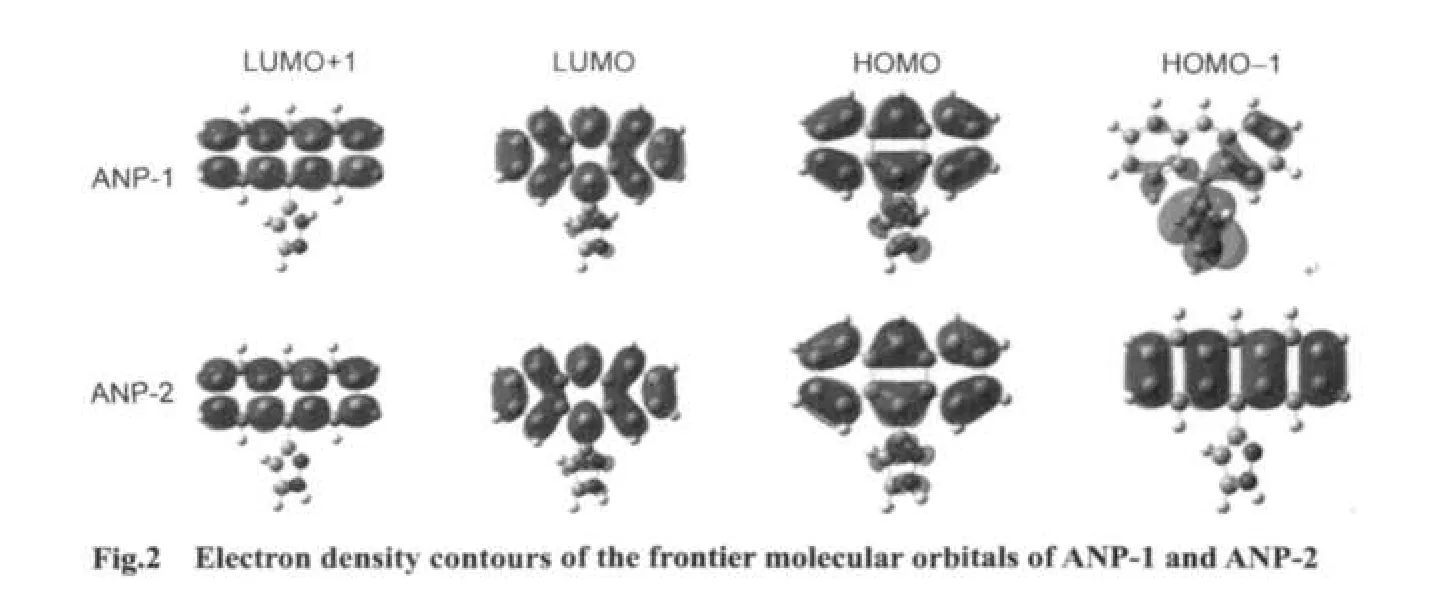
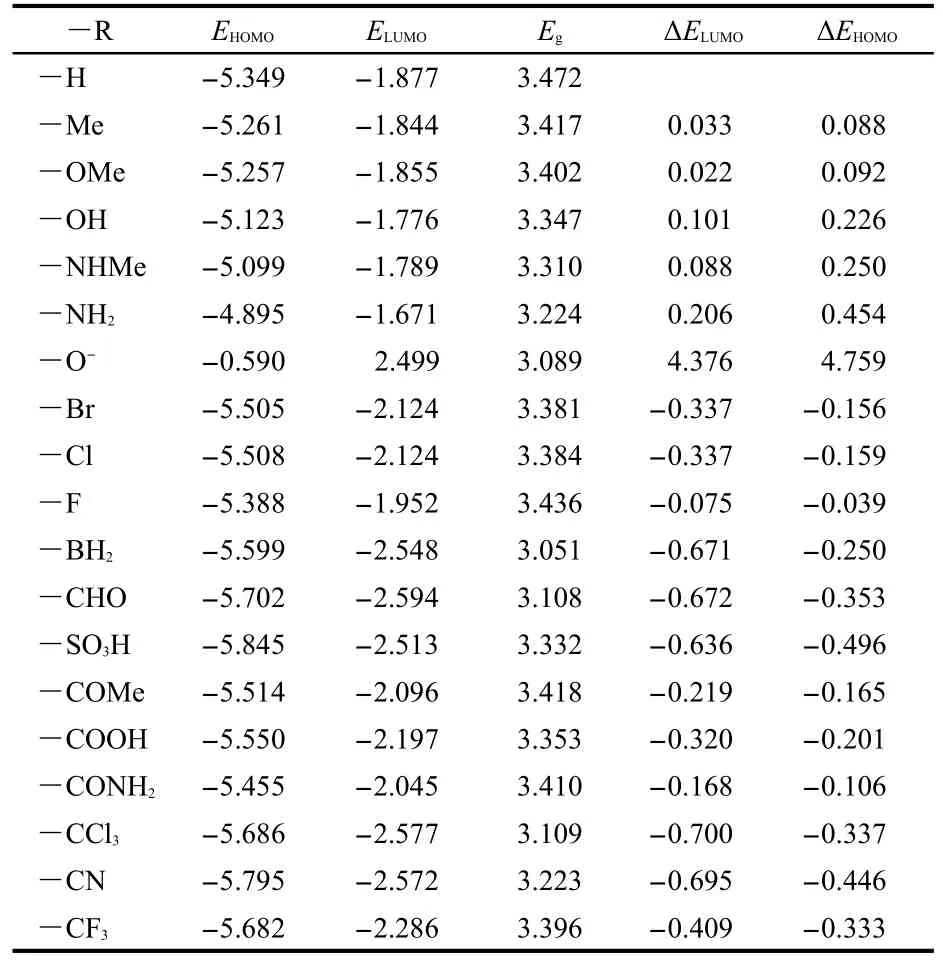
Table 2 Frontier molecular orbital energies(in eV)and their differences(Egin eV)obtained at the B3LYP/6-31G(d)level
3.2.3 Emission spectra
The calculated emission parameters are listed in Table 4.It can be seen from Table 4 that the fluorescence emission spectra of the derivatives are all red shifted compared with the parent molecule ANP-1,whatever the substituent is an electronwithdrawing group or an electron-donating group,consistent again with the FMO analysis above.Furthermore,the redshifted wavelength is predicted in the increasing order―Me<― OMe<― OH<― NHMe<—NH2for the electron-donating group,and ―CONH2<―COOH≈—CF3≈―CN≈―COMe<―SO3H<―CHO<―BH2<―CCl3for the electron-withdrawing group.Table 4 shows that the derivative with―R=―NH2possesses a relatively longer fluorescence emission wavelength(514 nm)than the derivative with―R=―NHMe(490 nm),the derivative with―R=―OH possesses a relatively longer emission wavelength(486 nm)than the derivative with―R=―OMe(472 nm),the derivative with―R=―CHO possesses a relatively longer emission wavelength(498 nm)than the derivative with―R=―COMe(478 nm),showing that the methyl group is not a good candidate for designing an optical material possessing a longer fluorescence emission wavelength.Table 4 also shows that among the derivatives with―R=―F,―Cl,―Br,the derivative with―R=―Br shows the longest fluores-cence emission wavelength(471 nm)whereas the derivative with―R=―F shows a shortest fluorescence emission wavelength(468 nm),suggesting that the electronegativity may play a role:a more electronegative group may result in a shorter emission wavelength,or,in another word,a more electropositive group may result in a longer emission wavelength.This is further confirmed by the fact that the derivative with―R=―CCl3has a longer fluorescence emission wavelength(531 nm)than the one with―R=―CF3(475 nm).Among the substituents considered in this work,we find that the derivatives with ―R=―O-,―BH2,―CCl3,―CHO,―NH2have relatively the longest fluorescence emission wavelength,suggesting that if one wants to design and synthesize an optical material possessing a longer fluorescence emission wavelength,one can substitute the hydrogen atom of the parent molecule with these groups.
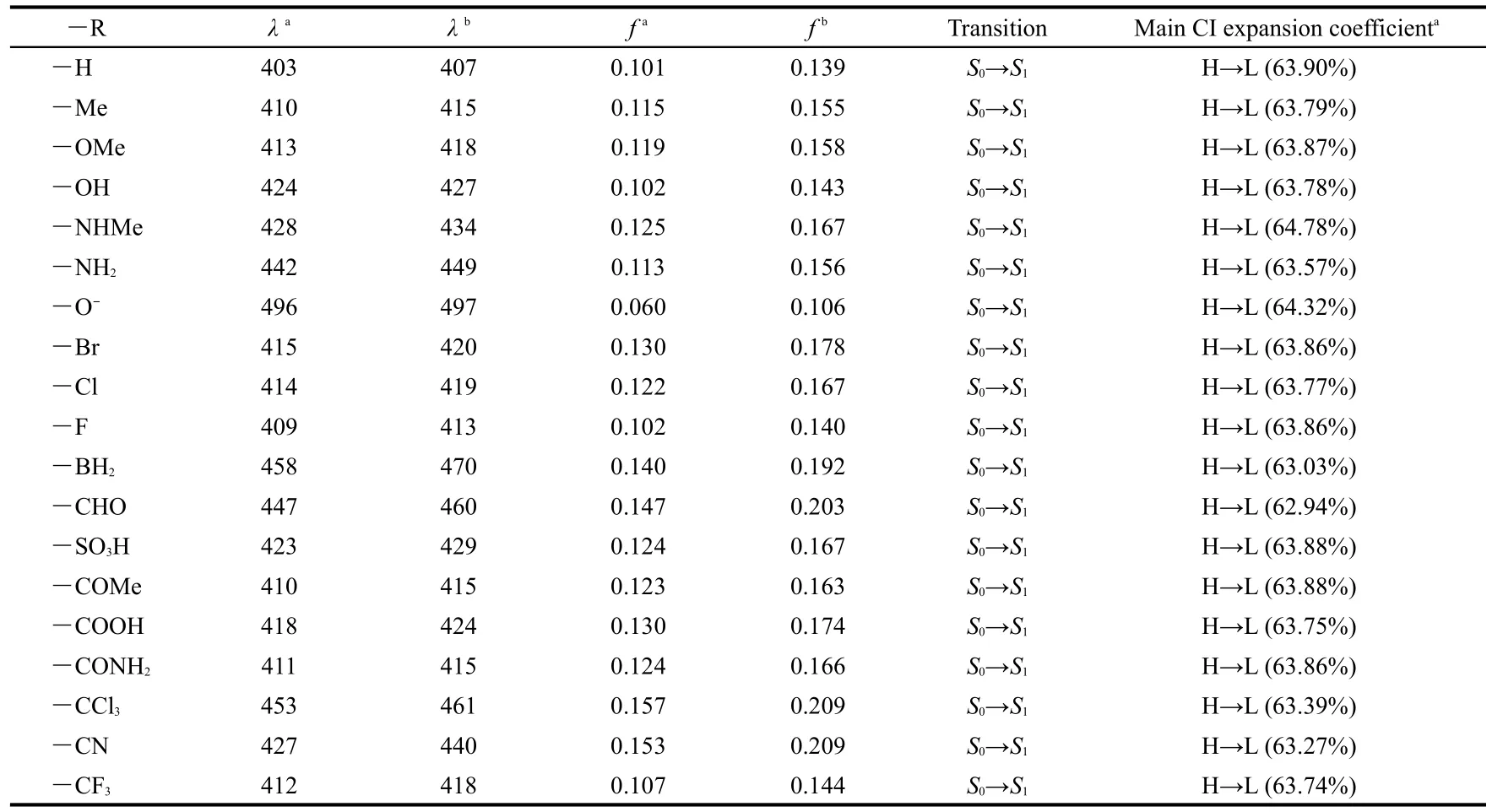
Table 3 Absorption wavelengths(λin nm),oscillator strengths(f),transition assignment and main CI expansion coefficients ofANP-1 and its derivatives
The data in the third column of Table 4 are the fluorescence emission wavelength in CHCl3solvent.These data show that the solvent leads to a further red shift for the fluorescence emission wavelengths with the red-shifted extent 4-17 nm.Moreover,oscillator strengthsfin CHCl3solvent are all larger than the counterpart ones in gas phase,displaying that the fluores-cent emitting spectrum is strengthened in CHCl3solvent.
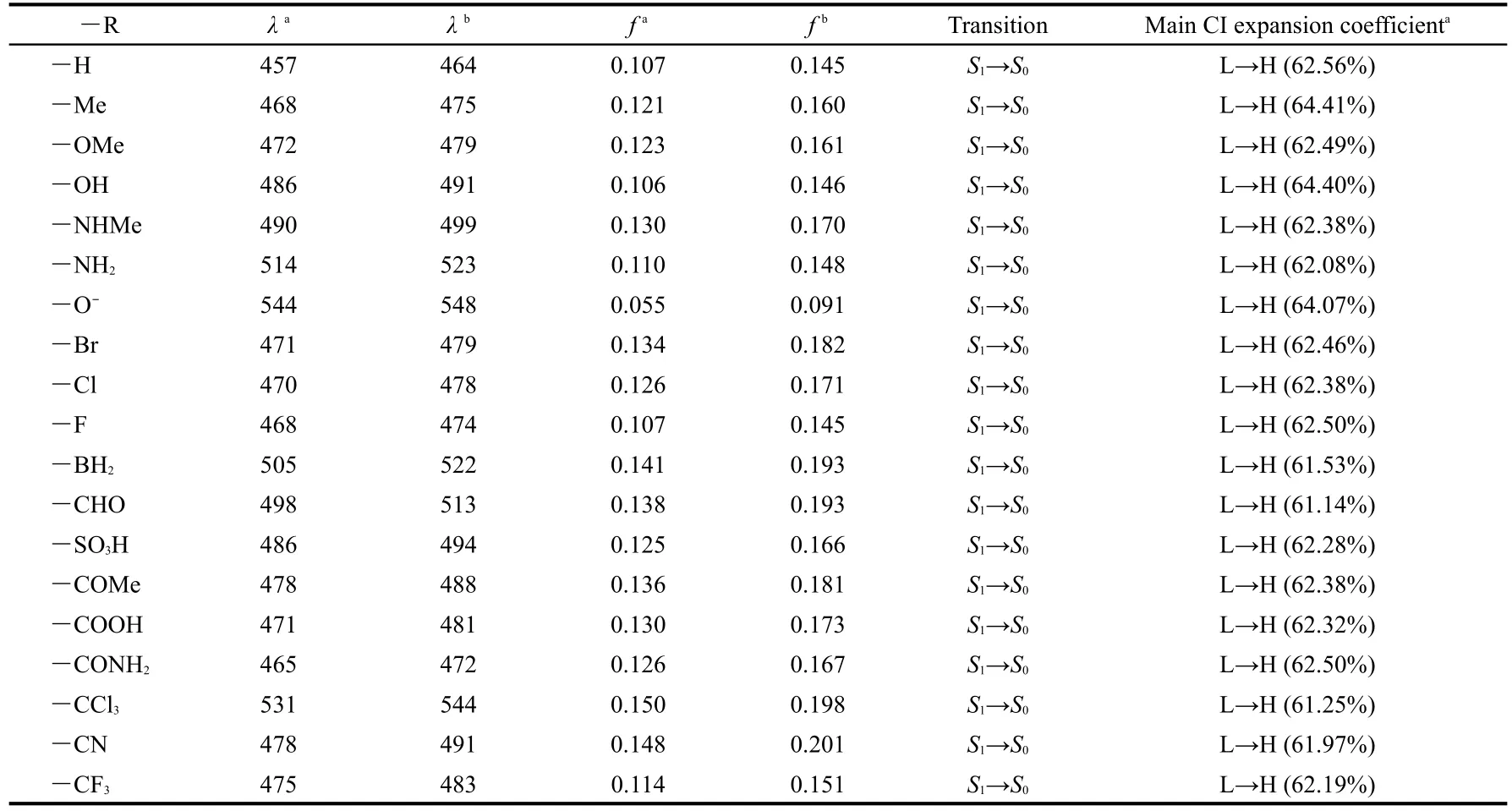
Table 4 Fluorescence emission wavelengths(λin nm),oscillator strengths(f),transition assignment and main CI expansion coefficients ofANP-1 and its derivatives
4 Conclusions
Based on the theoretical calculations we have demonstrated that,for all the derivatives of ANP considered in this paper,whatever the substituent is an electron-withdrawing group or an electron-donating group,the absorption and fluorescence emission wavelength values all show red shifts as compared with ANP.We have also shown that,compared with ANP,the derivatives of ANP-1 with ―R=―BH2,―CCl3,―CHO,and―NH2are good candidates both for the optical materials possessing longer absorption wavelength and for the optical materials possessing longer fluorescence emission wavelength.Furthermore,we found that the derivative with―R=―Br has both a longer absorption wavelength and a longer fluorescence emission wavelength than the derivative with―R=―F,and the derivative with―R=―CCl3has a longer wavelength than the one with―R=―CF3,showing that a more electropositive group may result in a longer absorption or emission wavelength.
(1)Mizukami,S.;Houjou,H.;Sugaya,K.;Koyama,E.;Tokuhisa,H.;Sasaki,T.;Kanesato,M.Chem.Mater.2005,17,50.
(2)Bader,M.M.;Custelcean,R.;Ward,M.D.Chem.Mater.2003,15,616.
(3)Wakamiya,A.;Ide,T.;Yamaguchi,S.J.Am.Chem.Soc.2005,127,14859.
(4) Murata,H.;Kafafi,Z.H.;Uchida,M.Appl.Phys.Lett.2002,80,189.
(5)Chen,J.;Law,C.C.W.;Lam,J.W.Y.;Dong,Y.;Lo,S.M.F.;Williams,I.D.;Zhu,D.;Tang,B.Z.Chem.Mater.2003,15,1535.
(6) Sapochak,L.S.;Benincasa,F.E.;Schofield,R.S.;Baker,J.L.;Riccio,K.K.C.;Fogarty,D.;Kohlmann,H.;Ferris,K.F.;Burrows,P.E.J.Am.Chem.Soc.2002,124,6119.
(7) Brinkmann,M.;Gadret,G.;Muccini,M.;Taliani,C.;Masciocchi,N.;Sironi.A.J.Am.Chem.Soc.2000,122,5147.
(8) Halls,M.D.;Schlegel,H.B.Chem.Mater.2001,13,2632.
(9)Geng,W.T.;Oda,M.;Nara,J.;Kondo,H.;Ohno,T.J.Phys.Chem.B 2008,112,2795.
(10)Shi,L.;Hong,B.;Guan,W.;Wu,Z.;Su,Z.J.Phys.Chem.A 2010,114,6559.
(11) Hu,B.;Gahungu,G.;Zhang,J.J.Phys.Chem.A 2007,111,4965.
(12) Sun,M.;Niu,B.;Zhang,J.Theor.Chem.Acc.2008,119,489.
(13)Fan,Y.;Zhao,Y.;Ye,L.;Li,B.;Yang,G.;Wang,Y.Crystal Growth&Design 2009,9,1421.
(14)Fan,Y.;Song,W.;Yu,D.;Ye,K.;Zhang,J.;Wang,Y.CrystEngComm 2009,11,1716.
(15) Gaal,M.;Gadermaier,C.;Plank,H.;Moderegger,E.;Pogantsch,A.;Leising,G.;List,E.J.W.Adv.Mater.2003,15,1165.
(16)Zhao,Y.;Gao,H.;Fan,Y.;Zhuo,T.;Su,Z.;Liu,Y.;Wang,Y.Adv.Mater.2009,21,3165.
(17) Gustafsson,G.;Cao,Y.;Treacy,G.M.;Klavetter,F.;Colaneri,N.;Heeger,A.J.Nature 1992,357,477.
(18) Chen,Y.;Au,J.;Kazlas,P.;Ritenour,A.;Gates,H.;McCreary,M.Nature 2003,423,136.
(19)Rakow,N.A;Suslick,K.S.Nature 2000,406,710.
(20) Zhang,H.;Zhang,Z.;Ye,K.;Zhang,J.;Wang,Y.Adv.Mater.2006,18,2369.
(21)Gao,L.;Lu,N.;Hao,J.;Hu,W.;Wang,W.;Wu,Y.;Wang,Y.;Chi,L.Langmuir 2008,24,12745.
(22)Gao,L.;Lu,N.;Hao,J.;Hu,W.;Shi,G.;Wang,Y.;Chi,L.Langmuir 2009,25,3894.
(23) Stephens,P.J.;Devlin,F.J.;Chabalowski,C.F.;Frisch,M.J.J.Phys.Chem.1994,98,11623.
(24) Foresman,J.B.;Head-Gordon,M.;Pople,J.A.;Frisch,M.J.J.Phys.Chem.1992,96,135.
(25)Cancès,E.;Mennucci,B.;Tomasi,J.J.Chem.Phys.1997,107,3032.
取代基對3(5)-(9-蒽基)吡唑光學(xué)性質(zhì)的影響
王昆鵬 王長生*
(遼寧師范大學(xué)化學(xué)化工學(xué)院,遼寧大連116029)
使用密度泛函理論(DFT)B3LYP/6-31G(d)方法優(yōu)化得到了3(5)-(9-蒽基)吡唑及其衍生物的基態(tài)(S0)分子結(jié)構(gòu),使用單激發(fā)組態(tài)相互作用(CIS)/6-31G(d)方法優(yōu)化得到這些分子的第一單重激發(fā)態(tài)(S1)的幾何結(jié)構(gòu),并使用含時密度泛函理論(TD-DFT)B3LYP/6-311++G(d,p)方法計算了它們的吸收和發(fā)射光譜.計算結(jié)果表明,與3(5)-(9-蒽基)吡唑相比,無論取代基是吸電子基團還是供電子基團,衍生物的吸收和發(fā)射峰均發(fā)生紅移,并且當(dāng)取代基―R=―BH2,―CCl3,―CHO,―NH2時衍生物有較長的吸收波長和發(fā)射波長.
吸收光譜; 熒光發(fā)射光譜;3(5)-(9-蒽基)吡唑; 激發(fā)態(tài)
O641
Received:October 29,2010;Revised:December 27,2010;Published on Web:January 18,2011.
?Corresponding author.Email:chwangcs@lnnu.edu.cn;Tel:+86-411-82159391.
The project was supported by the National Natural Science Foundation of China(20973088)and Research Fund of the Educational Department of Liaoning Province,China(2007T091,2008T106).
國家自然科學(xué)基金(20973088)和遼寧省高校創(chuàng)新團隊基金(2007T091,2008T106)資助項目
猜你喜歡
國際放射醫(yī)學(xué)核醫(yī)學(xué)雜志(2021年6期)2021-11-30 07:17:52
今日農(nóng)業(yè)(2021年2期)2021-11-27 19:19:53
今日農(nóng)業(yè)(2020年23期)2020-12-31 09:00:40
汕頭大學(xué)學(xué)報(自然科學(xué)版)(2020年4期)2020-12-14 07:04:58
汕頭大學(xué)學(xué)報(自然科學(xué)版)(2020年4期)2020-12-14 07:04:58
西安航空學(xué)院學(xué)報(2018年5期)2018-10-15 08:19:48
中國氣象科學(xué)研究院年報(2017年0期)2017-07-19 09:52:36
水利科技與經(jīng)濟(2016年2期)2016-04-09 02:09:13
合成化學(xué)(2015年1期)2016-01-17 08:59:30
原子與分子物理學(xué)報(2015年3期)2015-11-24 12:49:36
