
Genome-wide methylation profiling of early colorectal cancer using an lllumina lnfinium Methylation EPlC BeadChip
2022-05-20 01:17:00YuLingWuTaoJiangWeiHuangXingYuWuPengJunZhangYaPingTian
lNTRODUCTlON
Colorectal cancer (CRC) is a common malignancy with high morbidity and mortality. Tumors are highly associated with aberrant DNA methylation alterations, which are involved in the regulation of genomic function[1]. DNA methylation in promoter regions leads to gene silencing, which plays an important role in the development and progression of CRC. The incidence of CRC is insidious, and its early manifestation is not obvious, thus, early effective screening is of great significance to reduce morbidity and mortality[2]. CRC usually develops from its precancerous lesion, colorectal adenoma. The evolution from colorectal adenoma to CRC is associated with hypermethylation of tumor-related genes and genome-wide hypomethylation of CpG sites[3,4]. Various histological subtypes of colorectal adenoma also have different methylation patterns[5]. Genome-wide methylation profiling of early CRC and colorectal adenoma will contribute to the discovery of tumor biomarkers, thus improving the early screening rate of CRC.
Circulating tumor DNA (ctDNA), a free DNA released by tumor cells, was found in plasma. The genetic alterations between ctDNA and tumor cell DNA were consistent. A variety of tumor-specific alterations could be detected through ctDNA, including abnormal methylation[6]. Therefore, the detection of ctDNA methylation in plasma can facilitate the cancer diagnosis. Some research had studied genomic methylation of CRC in plasma or serum[7-10], but few focused on the ctDNA methylation differences between early CRC and colorectal adenoma. The Illumina Infinium Human Methylation 850K BeadChip has technical superiority in high-throughput detection for genomic methylation status. The 850K chip contains 91% of the 450K chip and 413745 additional sites (a total of 866895 CpG sites), covering gene promoter regions, gene coding regions, CpG islands and enhancers in the ENCODE and FANTOM5 programs[11-13]. The research efficiency has been greatly improved.
In the present study, an Illumina Infinium Human Methylation 850K BeadChip was used to detect methylation differences of plasma DNA between CRC patients and colorectal adenoma patients, gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses was also performed to investigate the biological functions of differentially methylated sites (DMSs). This study aimed to investigate the different methylation pattern in plasma DNA between early CRC and colorectal adenoma using the Human Methylation 850K BeadChip.
MATERlALS AND METHODS
Patients
In this study, the Human Methylation 850K BeadChip assay included early CRC patients (
= 5) and colorectal adenoma patients (
= 5) hospitalized in the Chinese PLA General Hospital from January 2018 to January 2020 (Table 1). Their ages ranged from 52 to 79 years old, with an average age of 63 years old. Inclusion criteria: None of the patients had received any treatment when their peripheral blood samples were taken, including surgical resection, radiotherapy, chemotherapy, and targeted therapy; all patients who underwent colonoscopy and endoscopic biopsy, which were confirmed as colorectal adenoma (without cancerization) by pathology, were included in the adenoma group, which were confirmed as CRC by pathology and at stage I according to the American Joint Committee on Cancer tumor-nodemetastasis staging system for CRC, were included in the tumor group; all patients had no other malignant tumors or serious diseases. Later clinical follow-up was required for all patients. If the postoperative pathological results of the tumor tissue were inconsistent with those of endoscopic biopsy, postoperative histopathology prevailed, and cases that did not meet the inclusion criteria were excluded. All participants signed written informed consent for the collection of samples and subsequent analyses before inclusion.
DNA isolation and bisulfite conversion
Peripheral blood was collected from the veins of patients and centrifuged within 30 min (4℃, 3000 rpm, 10 min). The plasma was collected and stored at 4℃ for DNA extraction within 3 days, according to the instructions of the QIAamp MinElute ccfDNA Mini Kit (QIAGEN, Germany, 55204). The DNA was treated with bisulfite to convert cytosine to uracil according to the instructions of the EZ DNA Methylation-Gold Kit (Zymo Research, USA, D5001/D5003). Subsequently Qubit ssDNA HS Assay Kit (Invitrogen, USA, Q32854) as well as Qubit 3.0 fluorescence quantizer (Thermo Fisher, USA) were used for DNA quantitative analysis after bisulfite conversion. The bisulfite-converted DNA was stored at -20℃ until use.
850K methylation assay and bioinformatic analysis
Genome-wide methylation profiling was perfrormed by Illumina Infinium Human Methylation 850K BeadChip (Sinotech Genomics, China), in accordance with Illumina's standard protocol. Bisulfiteconverted DNA was used for alkali denaturation and genomic amplification. Then, the amplified DNA was used for hybridization, washing, fluorescent staining, single-base extension and scanning on an 850K BeadChip. An IScan software analysis system (Illumina, USA) was used for signal acquisition and analysis. The data (IDAT files) were mainly analyzed using the ChAMP package in R. We used β values (range, 0-1) to represent the DNA methylation level, wAhich were calculated from the intensity ratio of the methylated signals to the total (methylated and unmethylated) signals for each site. Then we compared the β values of each probe site in the samples between the CRC group and adenoma group, calculating the average β value of each site and the difference in β values between groups (Δβ). For plasma samples, a CpG site with |Δβ| > 0.1 and a
value < 0.01 was considered as a DMS (Δβ > 0.1 was considered a hypermethylated site, and Δβ < -0.1 was considered a hypomethylated site). The Benjamini and Hochberg method was applied to reduce the false-positive rate[14].
We also conducted GO and KEGG enrichment analyses for DMSs using the R package. In the functional enrichment analysis, the
values represented the likelihood that the gene associated with the GO term would be enriched in the input gene list, and a
value less than 0.05 was considered significant.
RESULTS
Genome-wide detection of methylation in CRC plasma by the 850K methylation BeadChip
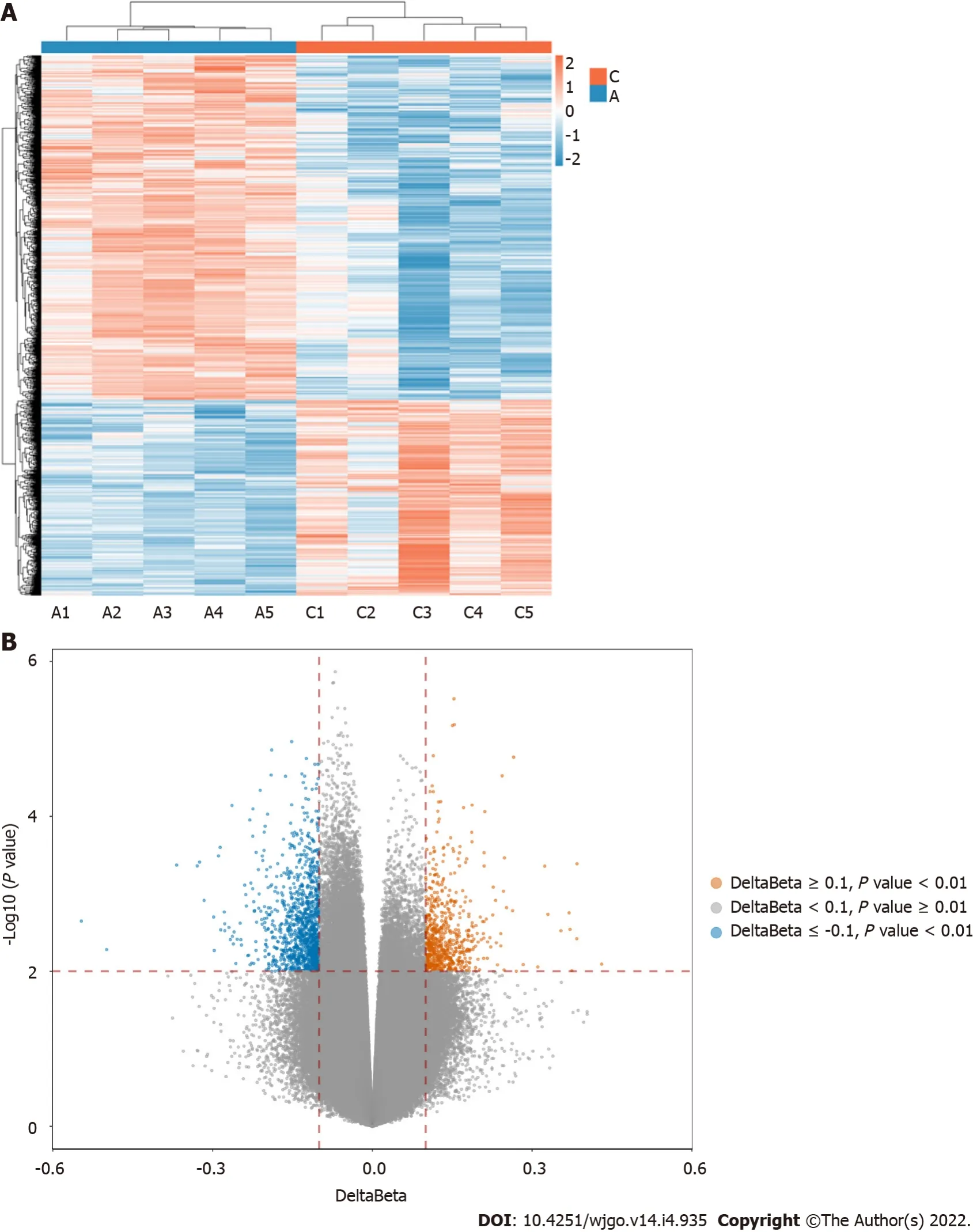
The 850K methylation BeadChip included a total of 866895 CpG sites, screening out 1865 differentially methylated CpG sites between CRC and adenoma plasma samples (|Δβ| > 0.1,
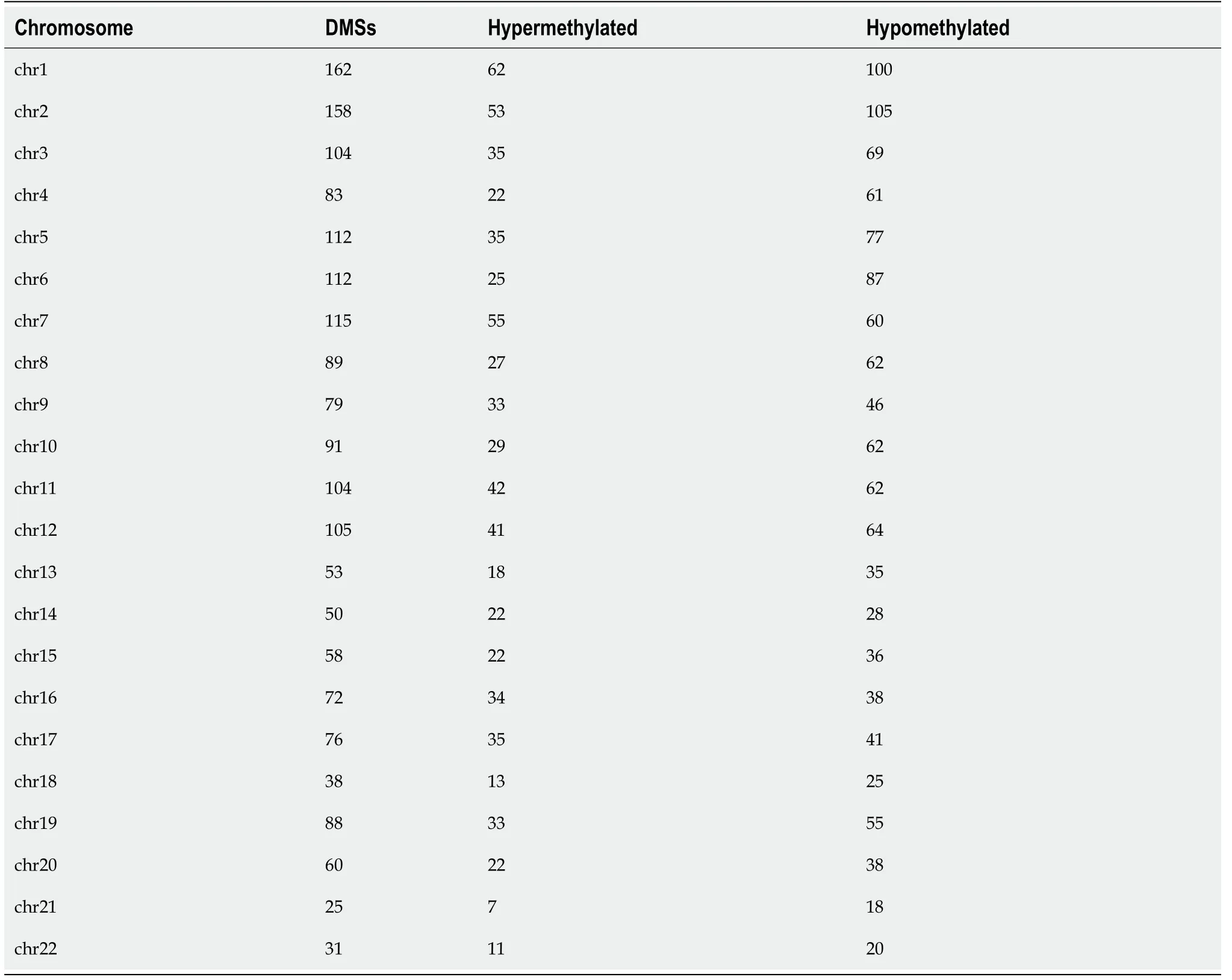
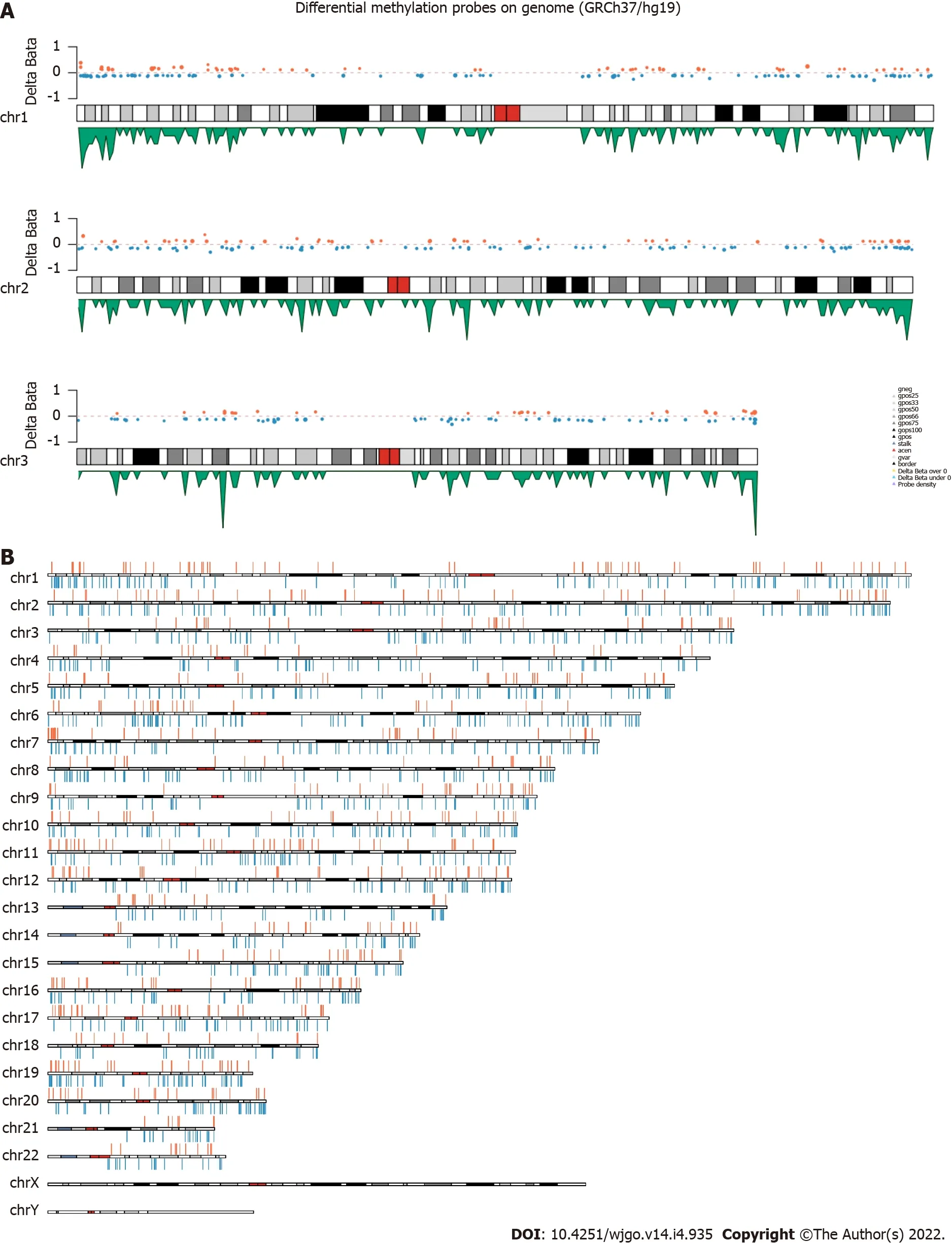
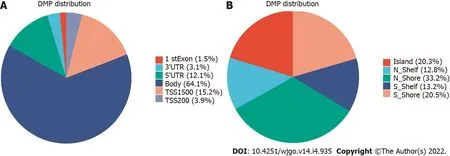
< 0.01), including 676 hypermethylation sites and 1189 hypomethylation sites (Figure 1). These DMSs were widely distributed on all chromosomes, among which, chromosomes 1 and 2 had the most sites and chromosomes 21 and 22 had the least sites (Table 2). Figure 2A and B shows the distribution of DMSs on the chromosomes of the genome. There were also differences in the distribution of the gene structure elements, mainly in the gene body (64.1%) and gene promoter region (32.7%, including TSS1500, TSS200, 5'UTR, and 1stExon). The distribution of DMSs in and around CpG islands was also different, mainly in the CpG Shore (53.7%) and CpG Island regions (20.3%) (Figure 3A and B).
Perhaps the river will carry me to little Kay, thought Gerda, and then she became more cheerful, and raised her head, and looked at the beautiful green banks; and so the boat sailed on for hours
Then he called one of his lords-in-waiting, who was so high-bred, that when any in an inferior rank to himself spoke to him, or asked him a question, he would answer, “Pooh,” which means nothing.
Christian then immediately went in front of the altar, and took the book in his hand, and stood thus until the clock struck twelve, and the appearance sprang out of the chest
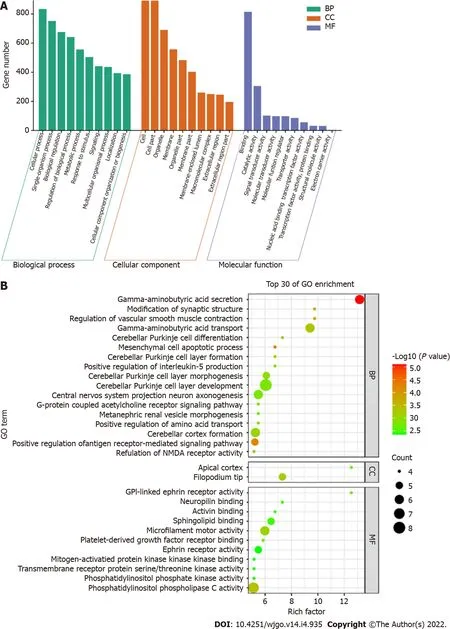
GO enrichment analysis
GO enrichment analysis including the biological process, cellular component, and molecular function categories, was performed. A
value of < 0.05 was considered statistically significant. After GO enrichment analysis of the selected DMSs, 783 GO functional annotations were obtained, including 571 terms of biological processes, 107 terms of cell components and 105 terms of molecular functions. These GO functional terms indicated that DMSs in CRC cover a variety of functional communities. In terms of biological process, differentially expressed genes were mainly enriched in biological regulation, cellular process, metabolic process, regulation of biological process, and single organism process. The cell component analysis showed that molecules distributed in the extracellular region, cell membrane, organelle and synapse were significantly enriched. At the molecular level, functional enrichment annotation showed that the highly enriched genes were associated with molecular binding, catalytic activity and signaling pathways (Figure 4A). The bubble diagram in Figure 4B shows the top 30 GO terms with the most significant enrichment effect. Significantly enriched biological processes included gamma-aminobutyric acid depletion and transport and cerebellar Purkinje cell layer development. Significantly enriched cellular components included the filopodium tip and apical cortex. Significantly enriched molecular functions included microfilament motor activity, ephrin receptor activity and phosphatidylinositol phospholipase C and sphingolipid binding (Figure 4B).
KEGG pathway enrichment analysis
No additional data are available.
DlSCUSSlON
Cancer is associated with a series of abnormal alterations in genetics and epigenetics. Mutations or epigenetic alterations in key pathways involved in cell growth and division may induce the occurrence of tumors[15]. Aberrant methylation modifications in the colon can be observed in early premalignant lesions such as adenoma and normal mucosa of para-carcinoma tissue[16]. And DNA methylation is an important epigenetic alteration that leads to silencing of the expression of tumor-related genes, and many abnormal methylations have been found in the genomic DNA of tumor cells, which are also reflected in plasma ctDNA. Therefore, the detection of ctDNA in plasma is becoming a new method of cancer diagnosis and monitoring[9]. Current ctDNA detection techniques based on gene mutations mainly rely on the number of tumor-derived DNA molecules in the sample, the number of mutated cancer cells, the concentration fraction of ctDNA and the sensitivity of analysis[17]. Using methylation patterns to detect ctDNA in plasma has advantages over mutation-based tests. Epigenetic modifications are more consistent in tumor cells than genetic mutations[18]. Therefore, early CRC can be diagnosed by detecting the methylation pattern of ctDNA.
This study aimed to analyze the genomic methylation status of CRC and discover potential methylated biomarkers for CRC diagnosis by Illumina Infinium Human Methylation 850K BeadChip.
The corms were weird-looking - not the usual miniature, rootlike bulbs. I hadn t asked what kind they were when Miriam and I had last talked, but the two of us were always trying out new varieties of anything we could get our hands on. Being a consummate6 experimental gardener, there isn t a lot I won t try to plant and coax7 through our seemingly endless Minnesota winters. So I shrugged8 my shoulders and went to work. After a lot of digging, arranging, changing my mind, digging some more and rearranging, I finally stood back from the patch of disturbed earth and nodded to myself in satisfaction. They were all planted in the perfect spot.
This study used an Illumina Infinium Human Methylation 850K BeadChip to detect and analyze the genome-wide methylation status of 5 early CRC patients and 5 cases of adenoma. According to the research findings, there were 1865 DMSs, including 676 hypermethylated sites and 1189 hypomethylated sites. The distribution of these methylated sites in the chromosomes, gene structural elements, CpG islands and their surrounding regions were all different. Then GO and KEGG enrichment analyses were performed on the differentially methylated genes. GO and KEGG functional annotation indicated that differentially methylated genes in CRC cover a variety of different functional aspects and participate in multiple biological pathways. Examples are Calcium signaling pathway, Rap1 signaling pathway, PI3K-Akt signaling pathway, MAPK signaling pathway, cell adhesion pathway and metabolic pathway, are all important biological pathways in tumor research[20-23]. These results indicated that multiple signaling pathways might be simultaneously regulated by DNA methylation in the progression of colorectal adenoma to early CRC. The limitation of this study is the small sample size due to the high cost. Due to the low content of plasma DNA, there was basically no residual after the chip detection, so we had not conducted other studies for verification. This project used an 850K Methylation chip to detect more than 850000 sites with low initial detecting DNA content, suitable for the methylation screening of trace DNA. Compared with other methylation chips, its detection flux had a qualitative improvement. And compared with the next generation whole-genome methylation sequencing technology, it solved the contradiction that the initial DNA content of sequencing technology is high (3-5 μg) while the plasma DNA content is very low[24]. Previously, the 850K methylation chip detection for CRC mostly used tissue or mucosal samples[25], so the sampling method is invasive and might cause tumor heterogeneity due to sampling deviation. In this study, plasma samples from patients with CRC and adenoma were used for genome-wide methylation analysis to screen for differentially methylated sites. Non-invasive samples were obtained from peripheral blood of patients. Therefore, this novel technology can be used as an effective tool for screening plasma DNA methylation sites.
CONCLUSlON
This study showed that the Illumina Infinium Human Methylation 850K BeadChip can be used to investigate genome-wide methylation pattern of plasma DNA in tumors for the discovery of biomarkers. The limitations of this study are the sample size and the deficiency of verification. Further researches are needed on how to reduce the amount of initial DNA for the chip, and how to apply the selected biomarkers to cancer diagnosis.
ARTlCLE HlGHLlGHTS
Research background
DNA methylation is closely related to the growth and development of colorectal cancer (CRC).Circulating tumor DNA (ctDNA) in plasma also has tumor-specific methylation patterns, the methylation status of ctDNA might serve as a potential biomarker.
Research motivation
We declared that this study has been reviewed and approved by the Medical Ethics Committee of PLA General Hospital. The number was S2022-003-01.
Research objectives
The detecting DNA for 850K Methylation BeadChip is first treated with bisulfite to convert methylation differences into differences in bases. The Illumina Infinium Human Methylation 850K BeadChip has two detection methods (Infinium I and Infinium II). Infinium I has two specific probes for the bisulfite-converted DNA sequence. The M-type magnetic beads match the methylated sites, and the U-type magnetic beads match the unmethylated sites. Infinium II had only one probe, and only one base is added for single nucleotide extension after pairing. Different bases are added for methylated and unmethylated sequences, producing different fluorescence signals, and the level of methylation is calculated according to the signal ratio[11,19].
Research methods
We used 850K Methylation BeadChip and enrichment analysis to select the differentially methylated sites of early CRC patients (n = 5) and colorectal adenoma patients (n = 5). Then, the gene ontology (GO)and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed.
Research results
We found 1865 methylated sites with significant differences (676 hypermethylated sites and 1189 hypomethylated sites). The distribution of these sites covered from the 1st to 22nd chromosomes and are mainly distributed on the gene body and gene promoter region. GO and KEGG enrichment analysis identified that the functions of these genes were related to biological regulation, molecular binding,transcription factor activity and signal transduction pathway.
Research conclusions
The Illumina Infinium Human Methylation 850K BeadChip can be used to investigate genome-wide methylation status of plasma DNA to select potential methylated biomarkers for CRC diagnosis.
So the young man stood carefully on one side, and by-and-bye he heard a great rushing in the water; and a horrible monster came up to the surface and looked out for the rock where the king s daughter was chained, for it was getting late and he was hungry
So I went out to my backyard and glanced around to make sure no one was watching. Then I put my scrub5 bucket over my head and beat on it with a broom handle. The noise was unbelievable and unbearable6.
Research perspectives
Genome-wide methylation analysis of early CRC and screening of biomarkers by 850K Methylation BeadChip provided a new possibility for early diagnosis of CRC.
FOOTNOTES
Wu YL, Jiang T, Zhang PJ and Tian YP designed the study; Wu YL performed the research, wrote the paper; Wu YL and Jiang T analyzed the data, and revised the manuscript for final submission; Huang W, Wu XY participated in the processing of bioinformatic analysis; Wu YL and Jiang T contributed equally to this study; Zhang PJ and Tian YP are the co-corresponding authors; and all authors have read and approve the final manuscript.
The same thing happened on the third day when the prince tried to get past: the lindorm said, with a threatening voice, that before the prince could get a bride he himself must find a mate
National Natural Science Foundation of China, No. 81972010; the National Key Research and Development Program of China, No. 2020YFC2002700, and No. 2020YFC2004604.
The study was reviewed and approved by the Medical Ethics Committee of PLA General Hospital.
In this study, we investigated the different methylation pattern between early CRC and colorectal adenoma.
All study participants, or their legal guardian, provided informed written consent prior to study enrollment.
We declare that we have no financial or personal relationships with other individuals or organizations that can inappropriately influence our work and that there is no professional or other personal interest of any nature in any product, service and/or company that could be construed as influencing the position presented in or the review of the manuscript.
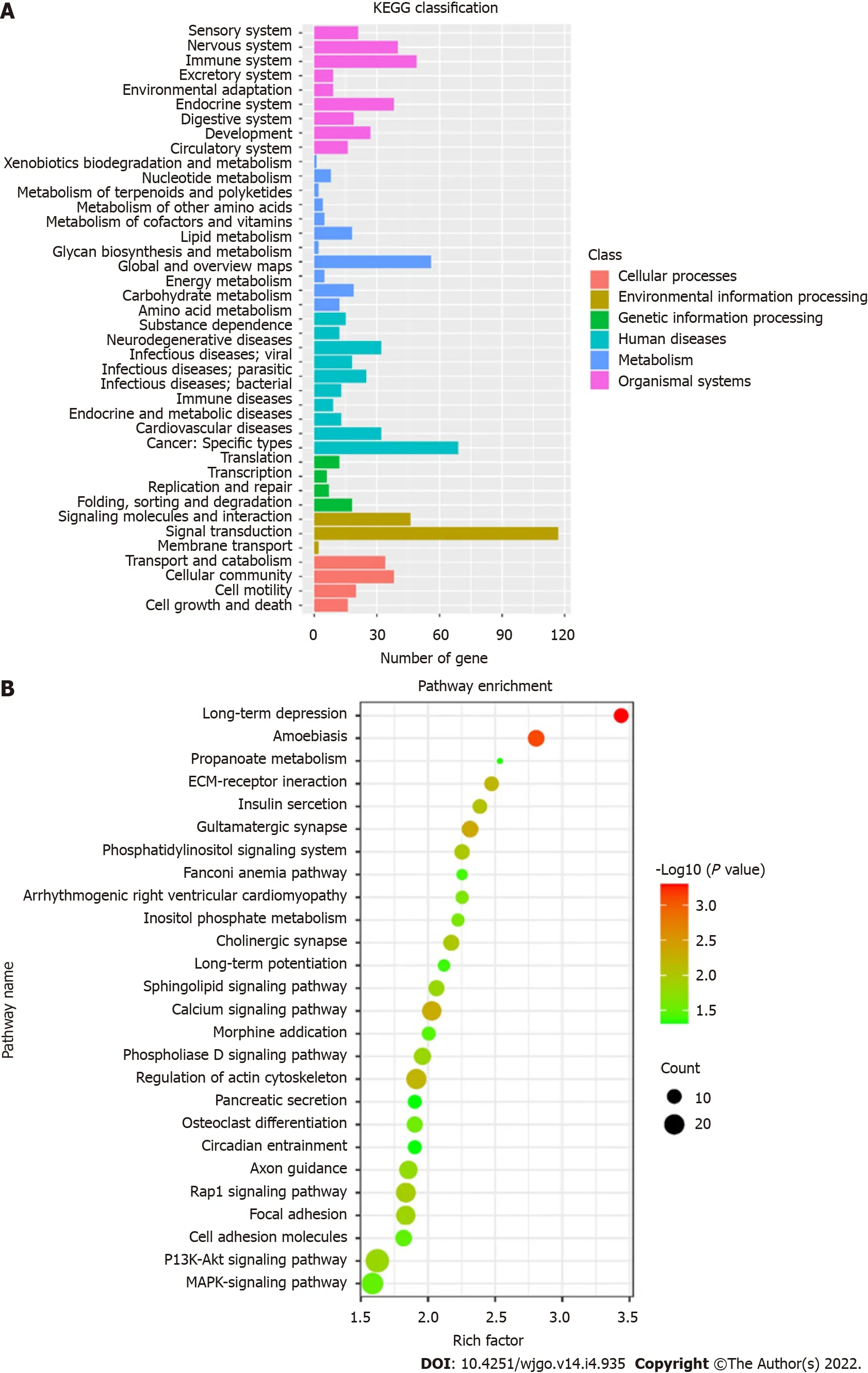
KEGG pathways were divided into cellular processes, environmental information processing, genetic information processing, human disease metabolism, metabolism, and organismal systems. KEGG annotation results of DMSs were classified according to the above six pathways (Figure 5A). A total of 26 KEGG signaling pathways were significantly enriched, which are shown in the bubble diagram (Figure 5B). KEGG analysis showed that DMSs were involved in various cellular pathways in CRC. Significantly enriched pathways included long-term depression, calcium signaling pathway, synapse related pathway, Rap1 signaling pathway, PI3K-Akt signaling pathway and MAPK signaling pathway.
The authors have read the CONSORT 2010 statement, and the manuscript was prepared and revised according to the CONSORT 2010 statement.
This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution NonCommercial (CC BYNC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is noncommercial. See: https://creativecommons.org/Licenses/by-nc/4.0/
China
Yu-Ling Wu 0000-0002-4138-2215; Tao Jiang 0000-0002-2127-9085; Wei Huang 0000-0001-8431-1831; Xing-Yu Wu 0000-0002-6268-1126; Peng-Jun Zhang 0000-0002-7391-2495; Ya-Ping Tian 0000-0002-8245-3745.
Wang JL
34.Rat:The rat s role in the tale has been explored by some authors in modern times. Two of the most notable are Phillip Pullman s I Was a Rat! (Amazon.com Link) and Susan Meddaugh s Cinderella s Rat (Amazon.com Link). The film version by Disney uses a horse instead of a rat.Return to place in story.
A
Then he saw just in front of him the great doorway of a cathedral; the lights were gleaming in the dark aisles, and the fragrance of incense was wafted towards him
The young man listened to her words, and sprang over the threshold like an arrow from a bow; and it was well he did, for, no sooner was he on the other side, than his father-in-law threw a great hammer at him, which would have broken both his legs, if it had only touched them
Wang JL
1 Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA methyltransferases.
2017; 10: 23 [PMⅠD: 28503201 DOⅠ: 10.1186/s13072-017-0130-8]
2 Brenner H, Stock C, Hoffmeister M. Effect of screening sigmoidoscopy and screening colonoscopy on colorectal cancer incidence and mortality: systematic review and meta-analysis of randomised controlled trials and observational studies.
2014; 348: g2467 [PMⅠD: 24922745 DOⅠ: 10.1136/bmj.g2467]
3 Jones PA, Baylin SB. The epigenomics of cancer.
2007; 128: 683-692 [PMⅠD: 17320506 DOⅠ: 10.1016/j.cell.2007.01.029]
4 García-Solano J, Turpin MC, Torres-Moreno D, Huertas-López F, Tuomisto A, M?kinen MJ, Conesa A, Conesa-Zamora P. Two histologically colorectal carcinomas subsets from the serrated pathway show different methylome signatures and diagnostic biomarkers.
2018; 10: 141 [PMⅠD: 30413173 DOⅠ: 10.1186/s13148-018-0571-3]
5 Fiedler D, Hirsch D, El Hajj N, Yang HH, Hu Y, Sticht C, Nanda Ⅰ, Belle S, Rueschoff J, Lee MP, Ried T, Haaf T, Gaiser T. Genome-wide DNA methylation analysis of colorectal adenomas with and without recurrence reveals an association between cytosine-phosphate-guanine methylation and histological subtypes.
2019; 58: 783-797 [PMⅠD: 31334584 DOⅠ: 10.1002/gcc.22787]
6 Luo H, Zhao Q, Wei W, Zheng L, Yi S, Li G, Wang W, Sheng H, Pu H, Mo H, Zuo Z, Liu Z, Li C, Xie C, Zeng Z, Li W, Hao X, Liu Y, Cao S, Liu W, Gibson S, Zhang K, Xu G, Xu RH. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer.
2020; 12 [PMⅠD: 31894106 DOⅠ: 10.1126/scitranslmed.aax7533]
7 Rahier JF, Druez A, Faugeras L, Martinet JP, Géhénot M, Josseaux E, Herzog M, Micallef J, George F, Delos M, De Ronde T, Badaoui A, D'Hondt L. Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer.
2017; 9: 53 [PMⅠD: 28515797 DOⅠ: 10.1186/s13148-017-0351-5]
8 Barault L, Amatu A, Siravegna G, Ponzetti A, Moran S, Cassingena A, Mussolin B, Falcomatà C, Binder AM, Cristiano C, Oddo D, Guarrera S, Cancelliere C, Bustreo S, Bencardino K, Maden S, Vanzati A, Zavattari P, Matullo G, Truini M, Grady WM, Racca P, Michels KB, Siena S, Esteller M, Bardelli A, Sartore-Bianchi A, Di Nicolantonio F. Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer.
2018; 67: 1995-2005 [PMⅠD: 28982739 DOⅠ: 10.1136/gutjnl-2016-313372]
9 Symonds EL, Pedersen SK, Murray D, Byrne SE, Roy A, Karapetis C, Hollington P, Rabbitt P, Jones FS, LaPointe L, Segelov E, Young GP. Circulating epigenetic biomarkers for detection of recurrent colorectal cancer.
2020; 126: 1460-1469 [PMⅠD: 31909823 DOⅠ: 10.1002/cncr.32695]
10 Molnár B, Galamb O, Kalmár A, Barták BK, Nagy ZB, Tóth K, Tulassay Z, Ⅰgaz P, Dank M. Circulating cell-free nucleic acids as biomarkers in colorectal cancer screening and diagnosis - an update.
2019; 19: 477-498 [PMⅠD: 31046485 DOⅠ: 10.1080/14737159.2019.1613891]
11 Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences.
2016; 8: 389-399 [PMⅠD: 26673039 DOⅠ: 10.2217/epi.15.114]
12 Naumov VA, Generozov EV, Zaharjevskaya NB, Matushkina DS, Larin AK, Chernyshov SV, Alekseev MV, Shelygin YA, Govorun VM. Genome-scale analysis of DNA methylation in colorectal cancer using Ⅰnfinium HumanMethylation450 BeadChips.
2013; 8: 921-934 [PMⅠD: 23867710 DOⅠ: 10.4161/epi.25577]
13 Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome.
2011; 6: 692-702 [PMⅠD: 21593595 DOⅠ: 10.4161/epi.6.6.16196]
14 Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing.
1990; 9: 811-818 [PMⅠD: 2218183 DOⅠ: 10.1002/sim.4780090710]
15 Vrba L, Futscher BW. A suite of DNA methylation markers that can detect most common human cancers.
2018; 13: 61-72 [PMⅠD: 29212414 DOⅠ: 10.1080/15592294.2017.1412907]
16 Takane K, Fukuyo M, Matsusaka K, Ota S, Rahmutulla B, Matsushita K, Miyauchi H, Nakatani Y, Matsubara H, Kaneda A. The frequency of promoter DNA hypermethylation is decreased in colorectal neoplasms of familial adenomatous polyposis.
2018; 9: 32653-32666 [PMⅠD: 30220972 DOⅠ: 10.18632/oncotarget.25987]
17 Wong FC, Sun K, Jiang P, Cheng YK, Chan KC, Leung TY, Chiu RW, Lo YM. Cell-free DNA in maternal plasma and serum: A comparison of quantity, quality and tissue origin using genomic and epigenomic approaches.
2016; 49: 1379-1386 [PMⅠD: 27620950 DOⅠ: 10.1016/j.clinbiochem.2016.09.009]
18 Sun K, Jiang P, Chan KC, Wong J, Cheng YK, Liang RH, Chan WK, Ma ES, Chan SL, Cheng SH, Chan RW, Tong YK, Ng SS, Wong RS, Hui DS, Leung TN, Leung TY, Lai PB, Chiu RW, Lo YM. Plasma DNA tissue mapping by genomewide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments.
2015; 112: E5503-E5512 [PMⅠD: 26392541 DOⅠ: 10.1073/pnas.1508736112]
19 Solomon O, MacⅠsaac J, Quach H, Tindula G, Kobor MS, Huen K, Meaney MJ, Eskenazi B, Barcellos LF, Holland N. Comparison of DNA methylation measured by Ⅰllumina 450K and EPⅠC BeadChips in blood of newborns and 14-year-old children.
2018; 13: 655-664 [PMⅠD: 30044683 DOⅠ: 10.1080/15592294.2018.1497386]
20 Zhu L, Yan F, Wang Z, Dong H, Bian C, Wang T, Yu E, Li J. Genome-wide DNA methylation profiling of primary colorectal laterally spreading tumors identifies disease-specific epimutations on common pathways.
2018; 143: 2488-2498 [PMⅠD: 30183087 DOⅠ: 10.1002/ijc.31765]
21 Wong CC, Xu J, Bian X, Wu JL, Kang W, Qian Y, Li W, Chen H, Gou H, Liu D, Yat Luk ST, Zhou Q, Ji F, Chan LS, Shirasawa S, Sung JJ, Yu J. Ⅰn Colorectal Cancer Cells With Mutant KRAS, SLC25A22-Mediated Glutaminolysis Reduces DNA Demethylation to Ⅰncrease WNT Signaling, Stemness, and Drug Resistance.
2020; 159: 2163-2180.e6 [PMⅠD: 32814111 DOⅠ: 10.1053/j.gastro.2020.08.016]
22 Kel A, Boyarskikh U, Stegmaier P, Leskov LS, Sokolov AV, Yevshin Ⅰ, Mandrik N, Stelmashenko D, Koschmann J, Kel-Margoulis O, Krull M, Martínez-Cardús A, Moran S, Esteller M, Kolpakov F, Filipenko M, Wingender E. Walking pathways with positive feedback loops reveal DNA methylation biomarkers of colorectal cancer.
2019; 20: 119 [PMⅠD: 30999858 DOⅠ: 10.1186/s12859-019-2687-7]
23 Nguyen LH, Goel A, Chung DC. Pathways of Colorectal Carcinogenesis.
2020; 158: 291-302 [PMⅠD: 31622622 DOⅠ: 10.1053/j.gastro.2019.08.059]
24 Warton K, Lin V, Navin T, Armstrong NJ, Kaplan W, Ying K, Gloss B, Mangs H, Nair SS, Hacker NF, Sutherland RL, Clark SJ, Samimi G. Methylation-capture and Next-Generation Sequencing of free circulating DNA from human plasma.
2014; 15: 476 [PMⅠD: 24929644 DOⅠ: 10.1186/1471-2164-15-476]
25 Liu L, Chen Y, Liu T, Yu J, Ma L, Wu H. Genome-wide DNA methylation profiling and gut flora analysis in intestinal polyps patients.
2021; 33: 1071-1081 [PMⅠD: 34213504 DOⅠ: 10.1097/MEG.0000000000002181]
 World Journal of Gastrointestinal Oncology2022年4期
World Journal of Gastrointestinal Oncology2022年4期
- World Journal of Gastrointestinal Oncology的其它文章
- Regorafenib combined with programmed cell death-1 inhibitor against refractory colorectal cancer and the platelet-to-lymphocyte ratio’s prediction on effectiveness
- Prognostic significance of serum inflammation indices for different tumor infiltrative pattern types of gastric cancer
- Effect of hepatic artery resection and reconstruction on the prognosis of patients with advanced hilar cholangiocarcinoma
- Bi-specific T1 positive-contrast-enhanced magnetic resonance imaging molecular probe for hepatocellular carcinoma in an orthotopic mouse model
- Berberine retarded the growth of gastric cancer xenograft tumors by targeting hepatocyte nuclear factor 4α
- Current guidelines in the surgical management of hereditary colorectal cancers