
Comprehensive molecular characterization and identification of prognostic signature in stomach adenocarcinoma on the basis of energy-metabolism-related genes
2022-02-25 06:51:08JinJiaChangXiaoYuWangWeiZhangCongTanWeiQiShengMiDieXu
INTRODUCTION
Gastric cancer (GC) is one of the most common malignancies in the digestive system.Within the last decades, the incidence rate of GC has gradually declined in some regions due to effective preventive measures and early diagnosis strategies[1].However, inoperable GC cases that are diagnosed at an advanced stage still have a poor prognosis[2]. According to the data of GLOBOCAN 2018, GC ranked third in global cancer mortality rate, only behind lung cancer and colorectal cancer in both genders combined[3]. Therefore, there is still urgent need to accurately predict the clinical outcomes of GC patients for the sake of more individualized management.
Reprogrammed metabolic pattern has long been recognized as a hallmark of cancers. Tumor cells can have different manners of nutrient acquisition and consumption compared to normal cells to obtain and maintain malignant features[4].The most well-known feature of cancer metabolism is the increased glycolysis and lactate production even in an oxygen-rich microenvironment, which is termed as“Warburg effect”[5]. Until now, it was generally believed that glucose is the major source of energy for cancer cells[6]. However, there is a growing awareness that the metabolic phenotype of cancer cells is largely heterogeneous. Some tumor cells primarily utilize glycolysis, while some other tumors have a metabolic property of oxidative phosphorylation (OXPHOS)[7]. Accumulating evidences show that there is a metabolic symbiosis between glycolysis and OXPHOS pathways in tumor cells[8]. For example, the lactate and pyruvate produced by glycolysis can act as the substrates of intermediates in the tricarboxylic acid cycle (TCA) to help generate adenosine triphosphate (ATP) in neighboring cells[9]. Similarly, some other non-glucose nutrients (free fatty acids, amino acids) may serve as the alternative fuels to fulfill the energy burden of tumor cells[10]. Since the complex metabolic characteristics of tumor cells can greatly influence the clinical fate of malignancies, a deeper understanding of the cancer metabolic fingerprint may be crucial to develop new therapies and identify promising prognostic signatures.
Everything went well for a week or a fortnight, and then the woman said, Hark you, husband, this cottage is far too small for us, and the garden and yard are little; the Flounder might just as well have given us a larger house. I should like to live in a great stone castle; go to the Flounder, and tell him to give us a castle. Ah, wife, said the man, the cottage is quite good enough; why should we live in a castle? What! said the woman; just go there, the Flounder can always do that. No, wife, said the man, the Flounder has just given us the cottage, I do not like to go back so soon, it might make him angry. Go, said the woman, he can do it quite easily, and will be glad to do it; just you go to him.
In the present study, we aimed to select key prognostic factors of GC among the 587 energy metabolism genes, and construct a potential metabolism-related model for the survival prediction of GC patients. The model was trained and verified among a total of 339 GC samples from The Cancer Genome Atlas () Stomach Adenocarcinoma STAD) dataset and 300 tumor samples from the GSE62254 dataset of the Gene Expression Omnibus (GEO). Moreover, molecular classification of GC based on the expression of energy-metabolism-related genes was also conducted to decipher the underlying role of metabolism in GC.
MATERIALS AND METHODS
Data source and processing methods
The TCGA-STAD dataset and GSE62254 dataset were analyzed for signature identification. The “Level 3” RNA sequencing (RNA-seq) data and clinical characteristic information of STAD tumor samples were collected from the TCGA-STAD dataset using the gdc-client tool (https://portal.gdc.cancer.gov/). Gene IDs were converted into official gene symbols according to the Genome Reference Consortium Human Build 38 (GRCh38) assembly. Only genes with average Fragments Per Kilobase per Million (FPKM) value greater than zero in more than 70% samples were included for the analysis. The microarray gene expression profiles and patients’ clinical information of GSE62254 dataset was downloaded from Gene Expression Omnibus database (GEO,https://www.ncbi.nlm.nih.gov/geo/). Probes were mapped to gene symbols according to the corresponding platform file GPL570. The progression-free survival(PFS) period of each STAD patient from the two datasets was calculated, and samples with PFS less than 30 d were excluded from the analysis. A total of 639 STAD subjects were thus analyzed with 339 from TCGA-STAD dataset and 300 from GSE62254 dataset (Table 1).
The metabolic-related pathways were downloaded from Reactome (https://reactome.org/) and a total of 587 energy metabolism-related genes from 11 pathways were screened out for variate selection (Supplementary Table 1). Among the 587 genes,one gene was not offered in TCGA-STAD dataset, and the FPKM counts of 2 genes were zero. Eventually, two comprehensive matrixes combining the expression levels of the 584 genes and the clinical information of STAD patients from the two independent datasets were generated separately for further analysis.
Identification of molecular subtypes using non-negative matrix factorization algorithm
In the training set, univariate Cox regression analysis was firstly performed to identify prognosis-associated genes from the hub genes (< 0.05). To minimize overfitting, least absolute shrinkage and selection operator (Lasso) regression analysis was further conducted using the R package glmnet for model construction[20,21]. The optimal lambda value was determined through 10-fold cross-validation. The coefficients of the variates included in the constructed model were estimated by the analysis and used to calculate the risk score of each STAD patient. Z-score normalization of risk score was further performed and zero was set as the cut-off value to determine the high-risk and low-risk patients.
Evaluation of immune characteristics between molecular subtypes
The enumeration of six tumor-infiltration immune cells (B cell, CD4T cell, CD8T cell,neutrophil, macrophage, neutrophils and dendritic cell) was estimated using the“Tumor Immune Estimation Resource” (TIMER, https://cistrome.shinyapps.io/timer/) tool[15]. The “Estimation of STromal and Immune cells in MAlignant Tumorsusing Expression data (ESTIMATE)” algorithm was applied to calculated the ImmuneScore and StromalScore which represent the relative proportion of immune cells and stromal cells in tumor tissues[16]. The ESTIMATEScore is the sum of ImmuneScore and StromalScore and refers to the purity of tumor tissues.
Since protein-protein interaction (PPI) analysis can help identify hub genes with core functions, PPI among genes in the identified key modules was further explored. The Search Tool for the Retrieval of Interacting Genes (STRING) is a well-known database containing comprehensive PPI information (version 11.0, https://string-db.org/). The PPI network among these genes was thus mapped to the STRING assembly and then visualized by the Cytoscape software. Important nodes in the network were identified by the Cytoscape plugin[19]. The topological analysis method Degree and the centrality analysis methods Closeness and Betweenness were used separately to identify the hub nodes in the PPI network. Among the top 15 hub nodes identified by each method, only genes with consistent high Degree, Closeness, and Betweenness values (larger than the median value) were selected as hub genes.
No! the poor little ones are playing with light, childishspirits. Play on, play on, thou little maiden9! Soon the years willcome- yes, those glorious years. The priestly hands have been laidon the candidates for confirmation10; hand in hand they walk on thegreen rampart. Thou hast a white frock on; it has cost thy mother much labor11, and yet it is only cut down for thee out of an old largerdress! You will also wear a red shawl; and what if it hang too fardown? People will only see how large, how very large it is. You arethinking of your dress, and of the Giver of all good- so glorious isit to wander on the green rampart!
Identification of hub genes by protein-protein interaction analysis
Network construction, hierarchical clustering analysis was performed firstly to remove the outlier samples. As previously described[17], the connection strength between each pair of genes (nodes in the network) was calculated by Pearson correlation analysis. The soft-threshold powerwas set to 8 in order to satisfy a scalefree topology with> 0.8. The topology overlapping matrix (TOM) was then constructed from the adjacency matrix to avoid the influence of noise and spurious associations. On the basis of TOM, average linkage clustering using the dynamic tree cut method was subsequently conducted to define co-expression modules. The size of genes in a module should be more than 30. Module eigengenes were further calculated to explore the relationship among modules. Modules with highly correlated eigengenes were merged together and eventually formed a new module network. The cut-off values of module integration parameters were set as height = 0.25, deepSplit =2, minModuleSize = 30. In order to identify the modules of interest, the correlation between each co-expression module and patients’ clinical features as well as cluster subtypes was further evaluated. Modules with a significant correlation to the energymetabolism subtypes of STAD patients were defined as key modules for the subsequent selection of hub genes (correlation coefficient >0.4,< 0.05).Functional enrichment analysis of genes in the key modules was further conducted using R package clusterProfiler[18].
5 pound for the fees and receive a short but heart-touching note in all women s eyes: I have no idea the clear rights and responsibilities for my left hand to my right hand, my right leg to my left leg, my left eye to my right eye and my right sphere of brain to the left
In her wrath5 she seized Rapunzel s beautiful hair, wound it round and round her left hand,42 and then grasping a pair of scissors in her right, snip12 snap, off it came, and the beautiful plaits lay on the ground. And, worse than this, she was so hard-hearted that she took Rapunzel to a lonely desert place,43 and there left her to live in loneliness and misery13.44
Construction and evaluation of identified prognostic signature
The factors in the potential prognostic model were selected from the hub genes identified by WGCNA and PPI analyses. Particularly, the 339 STAD samples in the TCGA-STAD dataset were randomly divided into two sets for the training (170)and testing (169) of the model (Table 1). In order to avoid selection bias, 100 times repetition sampling were conducted to the ensure the even distribution of patients’clinical characteristics between the training and testing sets. The Chi-squared test was performed and two-sidedvalues > 0.05 for all the parameters were considered to be efficient.
The non-negative matrix factorization (NMF) approach was applied for clustering analysis based on the gene expression data of TCGA dataset[11]. Firstly, univariate Cox regression analysis was conducted to identify survival-associated genes among the 584 energy metabolism-related genes. Then, the NMF algorithm and 50 runs were performed with the standard “brunet” pattern using the R package NMF[12]. The range of cluster number (value) was set as 2 to 10, and the minimum number of members per subtype was set to 10. The optimal number of k value was determined by several parameters including the cophenetic correlation coefficient[11], dispersion and silhouette[13] and residual sum of squares (RSS)[14] to ensure a robust clustering.
The nomogram integrating the identified signature and clinical information was built to improve the predictive capability[22]. The performance of the nomogram was assessed by calibration plot analysis. To assess the superiority of the identified energy metabolism-related prognostic signature, the predictive performance of the present model was further compared with the three other models proposed by previous studies using K-M survival analysis, ROC curve analysis, Harrell's concordance index(C-index), and decision curve analysis (DCA)[23].
Gene set enrichment analysis
The predictive performance of the prognostic model was evaluated among the 339 STAD patients with varied clinical features in thedataset. The results of subgroup survival analyses revealed that the 6-gene signature could effectively discriminate high-risk and low-risk patients among the elder, both sexes, all stage,Lauren intestinal type and microsatellite instability-high (MSI-H) subgroups, which expanded its potential application (Supplementary Figure 3A). Univariate and multivariate Cox regression analyses were further performed to evaluate the clinical independence of the identified signature. It was proved that the calculated risk score could independently predict the PFS of STAD patients without the interference of other clinical parameters in the TCGA dataset (Supplementary Figure 3B).
A creative publicist was walking by the blind man and stopped to observe that the man only had a few coins in his hat. He put a few of his own coins in the hat, and without stopping to ask for permission() , took the sign, turned it around, and wrote a new message. He then placed the sign by the feet of the blind man, and left.
Statistical analysis
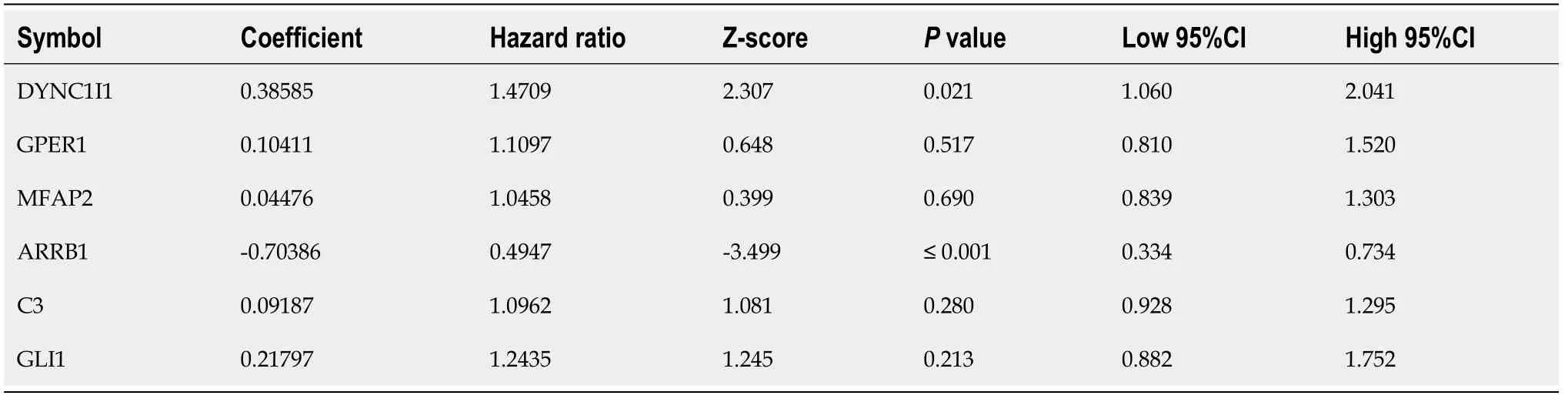
Among the six-energy metabolism-related genes, DYNC1I1, GPER1, MFAP2, C3 and GLI1 were risk factors while ARRB1 was a protective factor for clinical outcomes in GC. The prognostic value of the five risk genes have been sporadically reported in previous studies, while the protective value of ARRB1 in GC was rarely identified[44-49]. Functional enrichment analysis revealed that this metabolism-related signature was significantly involved in some classical cancer-related pathways. The interaction between the six genes and tumor metabolism and progression in GC deserves further investigation.
Every one was enchanted95, especially the prince, who called her his little foundling; and she danced again quite readily, to please him, though each time her foot touched the floor it seemed as if she trod on sharp knives
Then the Black Bird and the Green Giant resumed their natural form, for they were enchanters, and up flew Lolotte and Mirlifiche in their chariots, and then there was a great kissing and congratulating, for everybody had regained148 someone he loved, including the enchanters, who loved their natural forms dearly
RESULTS
Identification of molecular subtypes related to energy metabolism and cancer prognosis
Among the 584 EMRGs, a total of 86 genes were significantly associated with the prognosis of STAD patients according to the results of univariate Cox regression analysis with< 0.05 (Supplementary Table 2). The NMF analysis based on the expression of the 86 genes eventually identified two distinct subtypes (Cluster1 [123], Cluster2 [216]) among the 336 STAD patients in the TCGA dataset, which might have close association with cancer energy metabolism processes and prognosis in STAD (Figure 1A). Kaplan-Meier survival analysis revealed that the PFS of STAD patients of the two clusters was significantly different (= 0.025, HR = 0.66, 95%CI 0.46-0.95; Figure 1B). As shown in the heatmap of gene expression across the two clusters (Figure 1C), the great majority of EMRGs presented higher expression levels in Cluster 2 compared with Cluster 1.
Further comparison with World Health Organization classification suggested that Cluster 1 and Cluster 2 were inclined to the poorly cohesive and tubular subtypes,respectively (Figure 1D). Supported by the TCGA project, Adam[25] once divided STAD patients into foursubtypes:Epstein–Barr virus positive (C1), microsatellite unstable (C2), genomically stable (C3) and chromosomally unstable tumors(C4). By comparing the present results of molecular classification with the classicalfour subtypes-classification, we discovered that the identified Cluster 1 was inclined to the Epstein–Barr virus positive (C1) subtype which had a poorer prognosis,while Cluster 2 showed more relevance with the genomically stable (C3) subtype whose prognosis was much better (Figure 1E). Comparison analysis with other wellestablished clustering methods demonstrated the reliability of the classification results.
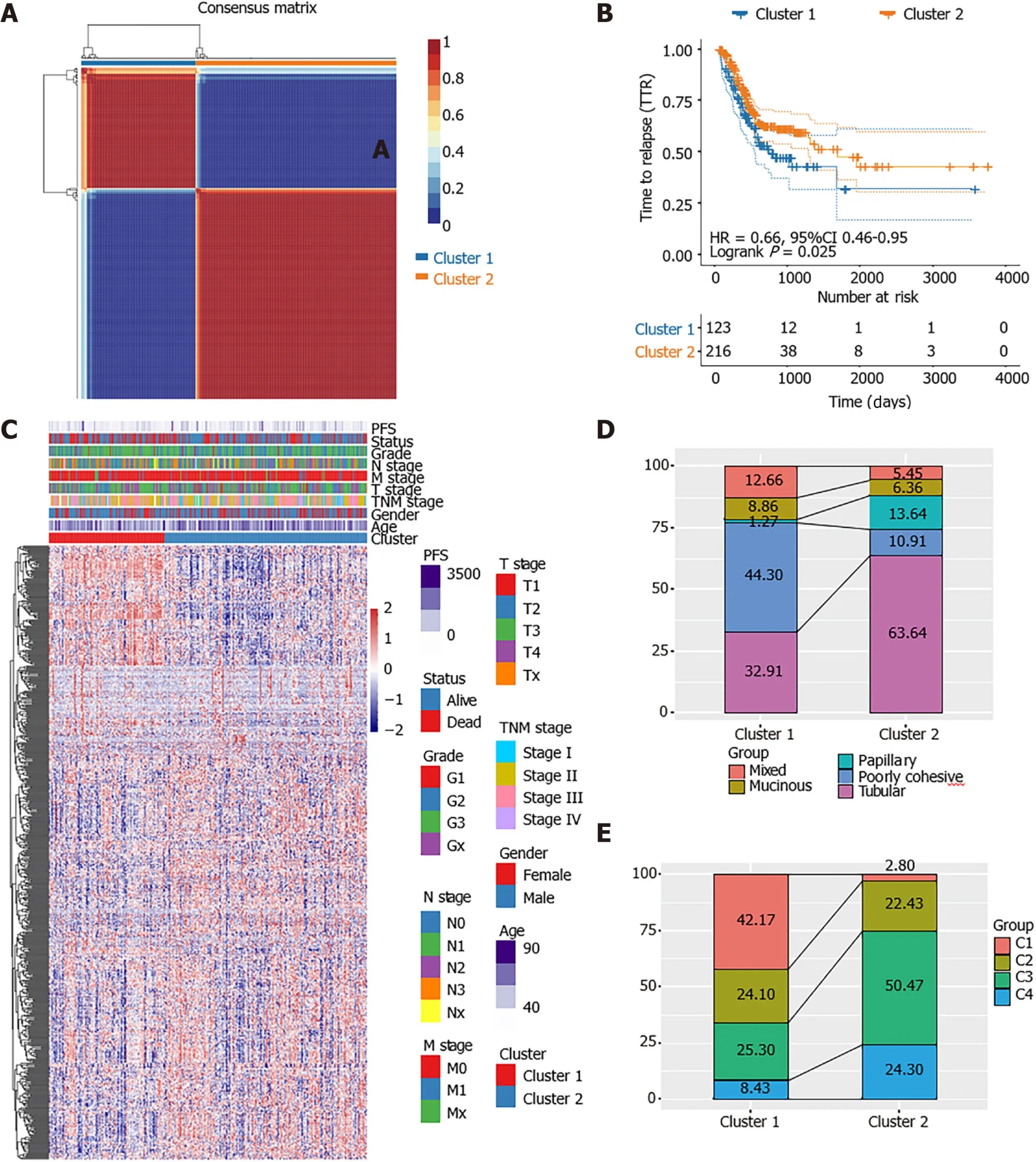
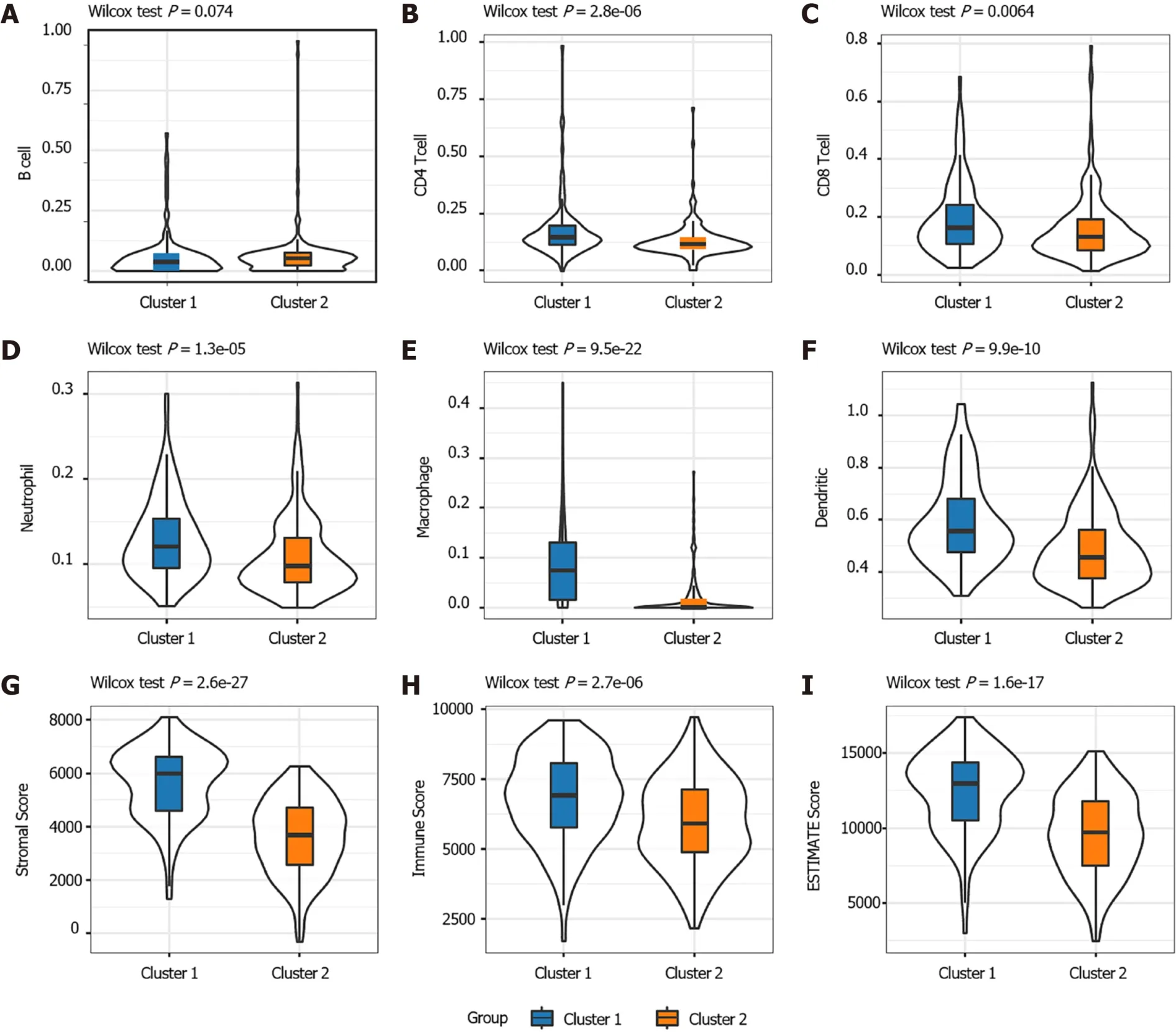
The distribution of the two clusters in STAD patients with different clinical characteristics was further analyzed. It was observed that most of patients with T1 or T2 stage and TNM Stage I were divided into Cluster 2 which had better survival. The Cluster 1 with poor outcomes inversely showed a trend of younger ages(Supplementary Figure 1). The proportions of tumor-infiltration immune cells and the fractions of immune and stromal cell components in tumor microenvironment (TME)were further compared between the two subgroups to explore the association between energy metabolism phenotype and immune status in STAD (Figure 2). The proportions of CD4T cells, CD8T cells, macrophage, neutrophils and dendritic cell were all significantly higher in Cluster 1 than in Cluster2 (Figure 2A-F). The calculated ImmuneScore, StromalScore, and ESTIMATEScore were also remarkably higher in Cluster 1, which represented more immune and stromal cell components in TME and lower tumor priority for the samples in Cluster 1 (Figure 2G-I). The results further suggested the close association between cancer cell energy metabolism, immune regulation, and clinical outcomes in STAD.
Selection of hub genes by WGCNA and PPI analyses
One outlier sample was identified by the hierarchical clustering analysis and removed from WGCNA co-expression analysis (Supplementary Figure 2A-C). Based on the expression of EMRGs in the TCGA dataset, a total of 29 co-expression modules were obtained after module fusion (Figure 3A, grey modules represent gene sets couldn’t be merged). The relationship between the identified modules and clinical characteristics as well as molecular classifications was shown in Figure 3B. It was concluded that Cluster 1 and Cluster 2 were significantly correlated with the yellow and brown module, respectively (> 0.4,< 0.05). The correlation between clinical phenotypes and the obtained modules as well as the genes of the modules was listed in Supplementary Table 3. As shown in Figure 3C, members in the yellow module were largely correlated with the Cluster 1 subtype, while members in the brown module were remarkably associated with the Cluster 2 phenotype. Therefore, the two modules having close relationship with energy metabolism-based subtypes of STAD were considered as the key modules, and the genes involved in these key modules were regarded as candidate genes for hub genes identification.
I sat down in a child-sized chair to catch my breath, hardly aware of what was happening, when she came to me with outstretched hands, bearing a small white box, unwrapped and slightly soiled, as though it had been held many times by unwashed, childish hands
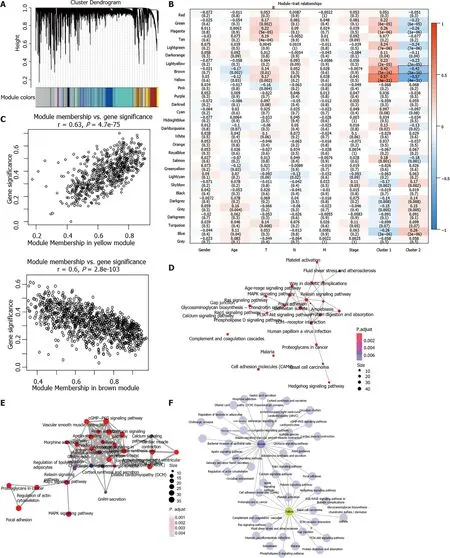
Functional enrichment analysis demonstrated that 23 KEGG pathways (MAPK signaling pathway, ECM?receptor interaction) were significantly involved in the yellow module (FDR < 0.01; Figure 3D, Supplementary Table 4) and 35 pathways (cGMP?PKG signaling pathway, Rap1 signaling pathway) significantly involved in the brown module (FDR < 0.01; Figure 3E, Supplementary Table 5). Most of these pathways were classical cancer-related biological processes. Moreover, the crosstalk of pathways was quite limited (Figure 3F), which further demonstrated the functional heterogeneity of the two key modules.
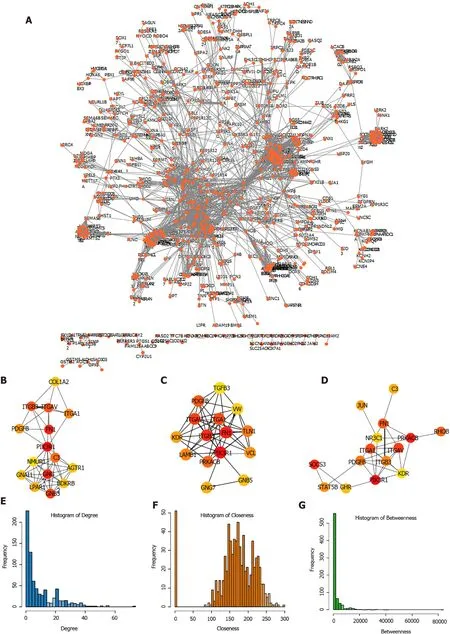
Subsequently, the expression of the candidate genes in key modules was mapped to STRING database to construct PPI network. A total of 3585 PPI pairs with a score higher than 0.9 were matched among the 1713 co-expression genes (Figure 4A,Supplementary Table 5). The top hub genes identified by the Degree (Figure 4B),Closeness (Figure 4C), and Betweenness (Figure 4D) methods were largely consistent(Supplementary Table 6). The topological properties of the PPI network were also evaluated and the distributions of degree, closeness, and betweenness were shown in Figure 4E-G. A total of 220 genes that satisfied high degree, closeness, and betweenness scores were selected out as hub genes for further analysis (Supplemen tary Table 7). These hub genes were assumed to be strongly correlated with the development of STAD, and were enrolled for subsequently identification of prognostic gene.
Identification of energy metabolism-related prognostic model
The clinical information of STAD patients in the training (170), testing (169)and external validation (300) sets used for model construction and evaluation was listed in Table 1. In the training set, after the selection of univariate Cox regression and Lasso regression analysis (Supplementary Figure 2D and E), six genes (,,,,and) out of the 220 hub genes were included in the prognostic model (Table 2). And a gene-based prognostic model was established to evaluate the survival risk of each patient as follows:Risk score = 0.38585 × exp +0.10411 × exp + 0.04476 × exp - 0.70386 × exp + 0.09187 × exp + 0.21797 ×exp.
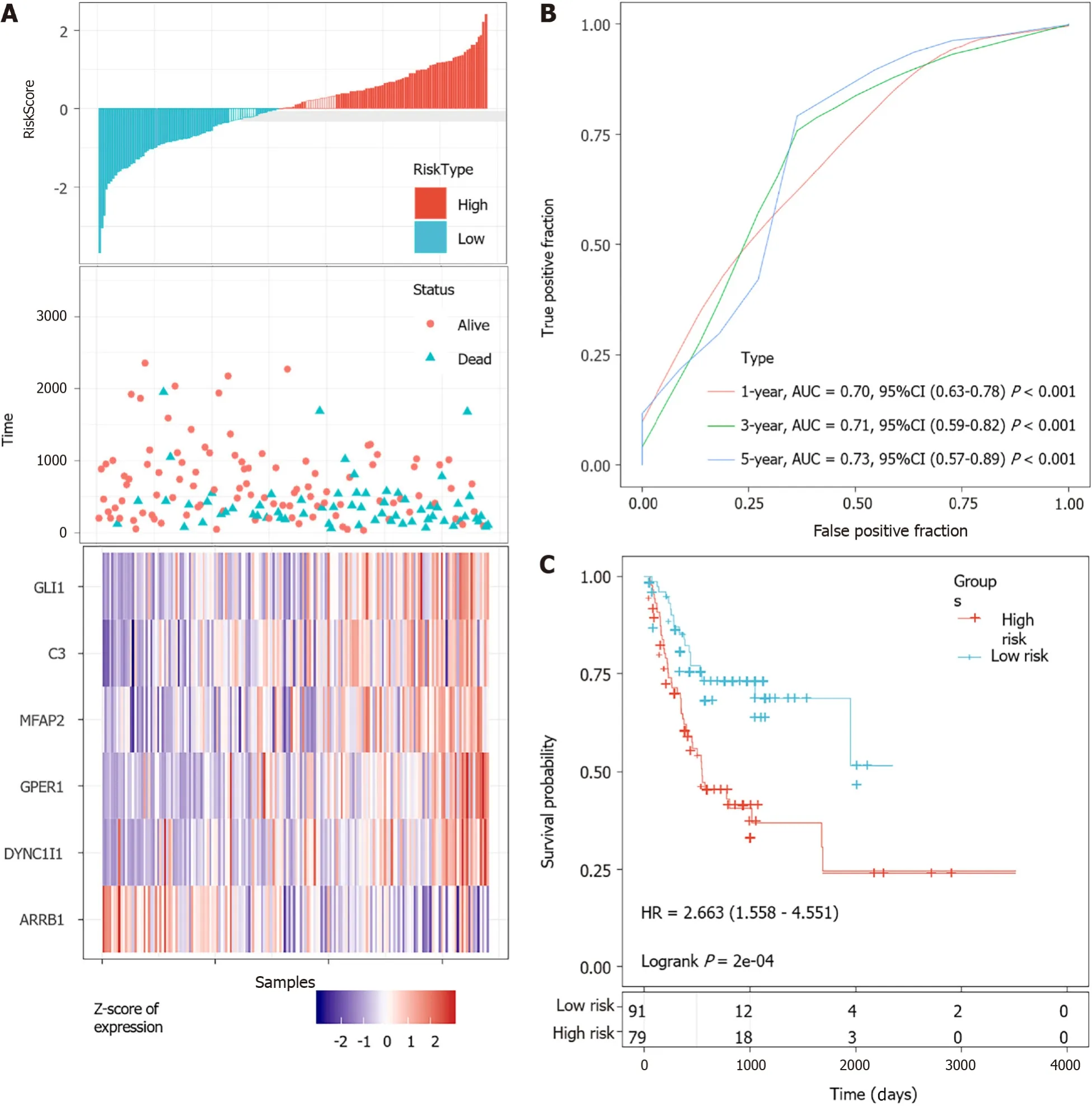
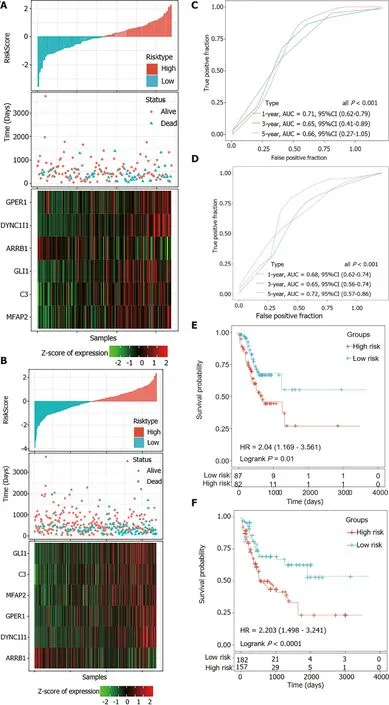
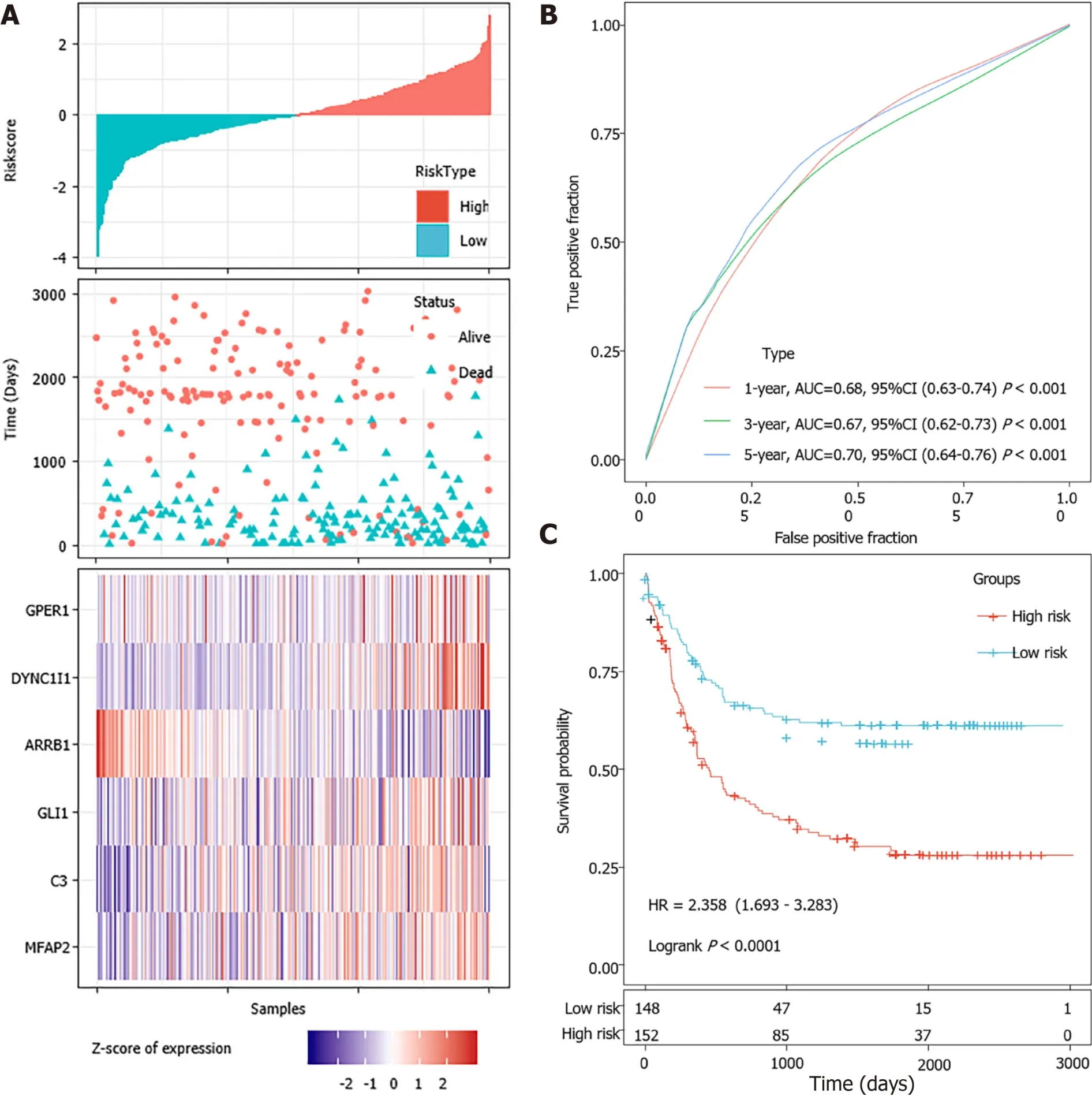
According to the cut-off value of normalized risk score (Z-score = 0), STAD patients were divided into high- and low-risk groups. The distribution of risk scores in the training set was shown in Figure 5A, which showed that expression levels of,,,, andwere positively correlated with risk scores, whilelevels was negatively correlated with risk scores. It was concluded that higherexpression was associated with a worse prognosis and was a favorable prognostic, while the other 5 genes were identified as unfavorable prognostic factors for STAD patients. The AUCs of 1-year, 3-year and 5-year ROC curves for the 6-gene signature to predict STAD survival were 0.70, 0.71 and 0.73, respectively (Figure 5B).Kaplan-Meier survival analysis confirmed that the high-risk group had significantly worse PFS than the low-risk group (Figure 5C).
24.Swear by heaven not to say a word about the matter: Promises, while important today, were more powerful in the past when honor was a great motivator. Also, before the time of literacy among the masses and written contracts, verbal promises were given greater weight. A promise was a contract and actionable by law if broken. Folklore emphasizes the importance of a promise by meting79 punishment upon those who do not keep their promises. In this story, the oath spares the princess life. Return to place in story.
A total of 300 STAD samples in GSE62254 were analyzed for the external validation of the signature. In this dataset,was a consistent protective factor while the other five genes were still risk factors for STAD survival (Figure 7A). The robustness of the signature was further verified (Figure 7B and C).
The wolf, the bear, and the wild boar arrived on the spot first, and when they had waited some time for the fox, the dog, and the cat, the bear said, I ll climb up into the oak tree, and look if I can see them coming
Association between the identified signature and clinical characteristics
Gene set enrichment analysis (GSEA) was performed to identify the functional difference between the high-risk and low-risk STAD patients in the TCGA dataset.Briefly, expression levels of all the protein-coding genes were input for analysis using the GSEA software (version 4.0.3). The classical gene sets of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (c2.cp.kegg.v7.0.symbols) were considered to decipher the phenotype. For each analytical pathway, the enrichment score (ES) and the significance of ES were calculated, and the normalized enrichment score (NES) and false discovery rate (FDR) were further calculated to examine functional enrichment results. An FDR cutoff value of 0.05 was considered in this test.
Comparison with previous prognostic models
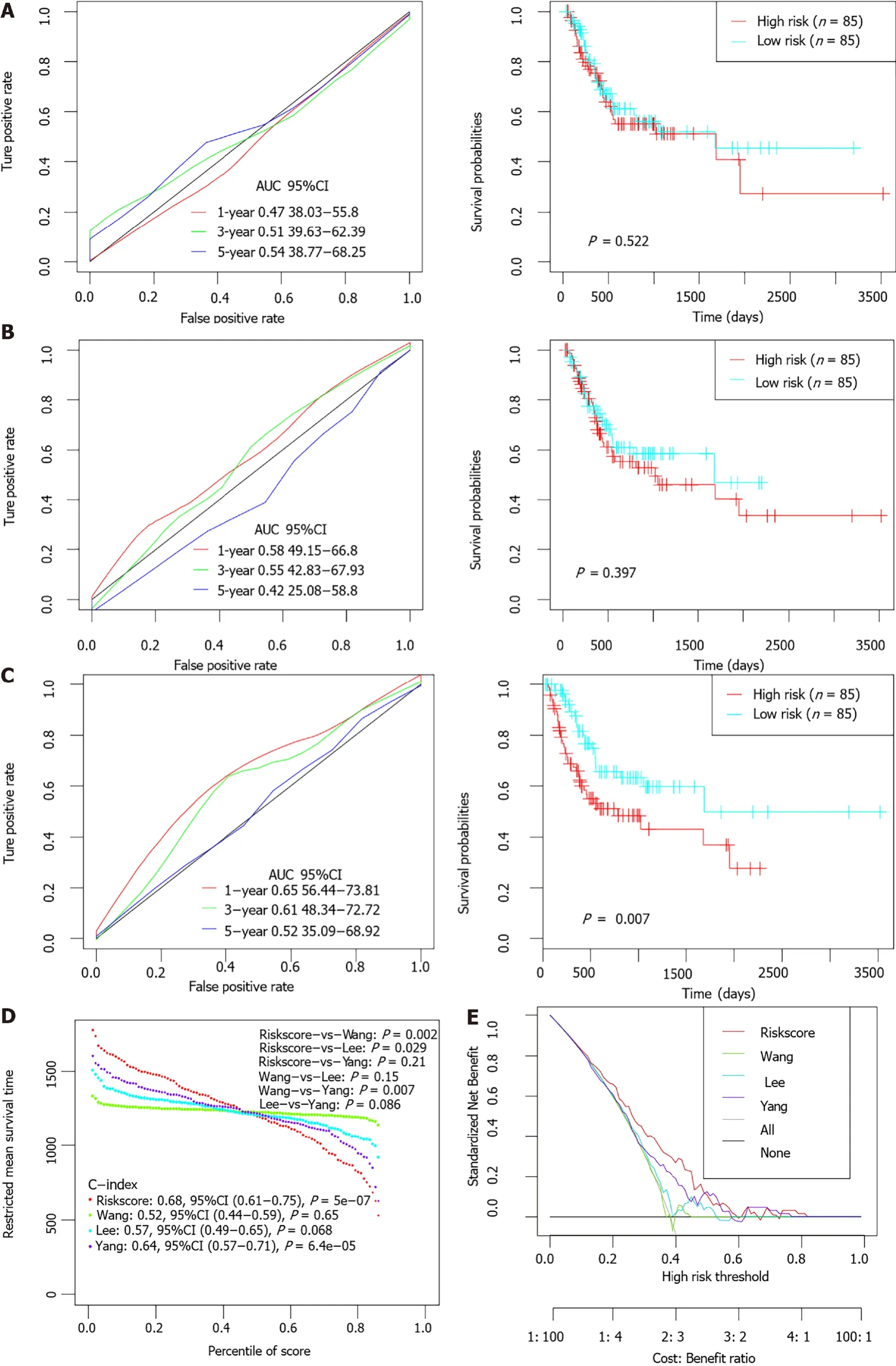
Previous studies had identified several prognostic models for survival prediction of STAD patients. The predictive performance of the present 6-gene signature was further compared with three previous models (a 5-gene signature proposed by Wang[26], a 6-gene signature proposed by Lee[27], and a 10 immune-related gene signature proposed by Yang[28]. For normalization, gene expression levels in each model were uniformly extracted from the original matrix of the-STAD dataset.The risk score of each STAD patient was calculated accordingly based on the corresponding coefficients provided by each model. Patients were divided into high-and low-risk groups separately according to the median value of normalized risk score for each signature. The comparative plots of Kaplan-Meier survival curve and ROC curves were shown in Figure 8A-C. Restricted mean survival time (RMST) was applied to calculated and compared the C-index of all signatures. The AUCs of the present 6-EMRG models were relatively higher and more stable than the other signatures, and the C-index was the highest among the four models (Figure 8D). DCA curves further demonstrated that the 6-gene signature had better clinical utility than the other models in the survival prediction of STAD patients (Figure 8E).
she let them down, and the Prince climbed up as usual, but instead of his beloved Rapunzel he found the old Witch, who fixed14 her evil,45 glittering eyes on him, and cried mockingly:
GSEA analysis of enriched pathway based on risk score
ssGSEA was performed to determine the potential related pathways according to patients’ prognostic risk in the entiredataset, and pathways with FDR < 0.05 were derived. By dividing samples into high-risk group and low-risk group based on whether the Riskscore is greater than 0, and analyzed the enriched pathway in both groups by using GSEA, we found that 10 pathways were significantly enriched in the high-risk group, such as ECM receptor interaction, hedgehog signaling pathway and; while only citrate cycle TCA cycle was significantly enriched in the low-risk group(< 0.05; Supplementary Figure 4). Thus, the 6-gene signature may be involved in the development and progression of STAD by participating in these pathways.
DISCUSSION
Cumulative evidence has revealed that metabolic reprogramming in cancer has extensive ties with oncogenesis and immune disorder[29,30]. In GC, previous studies suggested that the metabolic alteration in GC was typically characterized by increased glycolysis and repressed aerobic respiration for glucose metabolism, elevated consumption of some amino acids (especially glutamine) for amino acid metabolism,and upregulated fatty acid β-oxidation and oxidative degradation for lipid metabolism and others[31-33]. Moreover, there is a complex interplay among these reprogrammed metabolic pathways which forms the unique metabolic contexture in GC[34].
The detection of aberrant metabolomics also contributes to the identification of novel biomarkers for GC diagnosis or prognostic prediction, and the discovery of potential targets for GC treatment. For example, there are significant differences in metabolic profiles not only between GC patients and normal controls but also among different pathological GC subtypes, and the metabolic alterations have helped identify several promising biomarkers such as 3-hydroxypropionic acid and pyruvic acid in serum, phenylalanine in gastric juice, and alanine in urine[35-37]. Chen[38] once discovered proline and serine metabolites that could significantly discriminate metastatic animal models with GC from the non-metastatic samples. GC patients with higher levels of proline, p-cresol and 4-hydroxybenzoic acid in urine might have a worse prognosis according to a population-based study[39]. Taken together, the distinct features of energy metabolism in GC are worth investigating and may indicate novel biomarkers related to metabolism. However, the accurate detection of metabolites in biological samples is still hampered by some technical defects such as lack of optimized study methods, limited coverage in metabolomics fingerprints and interference caused by unwanted sources[40]. Moreover, the abundance of some metabolites can be quite low even less then the detection limit[41]. Gene expression profiling, with the advantage of being convenient and precise, can give a whole picture of tumor properties based on quantitative data[42]. By analyzing the expression levels of energy metabolism-related genes in GC tumor tissue, the metabolic characteristics of GC can be comprehensively interpreted from another dimension.
In the present study, a total of 587 energy metabolism-related genes were selected from the Reactome database. These genes are main participants in the key pathways of carbohydrate, protein, and lipid metabolism. Based on the expression data of the TCGA-STAD dataset, GC patients were divided into two metabolic subtypes using the NMF algorithm. Significant difference was observed in patients’ clinical characteristics and survival state between the two subtypes. This phenomenon further demonstrated the important role of energy metabolism in the development and long-term survival of GC. In addition, previous evidence has proved that metabolic interventions have crucial function in the modulation of cancer immunology[43]. In this study, when the tumor-infiltration immune cells and non-tumor components in TME were compared between the two groups, it could be observed that the proportion of almost all the immune cells and the fraction of immune components were significantly different between the two subtypes with varied metabolic features, which strongly indicated the close relationship between tumor metabolism and immunology in GC. Combined with findings from previous research, the results of this study confirmed the significance of identifying potential prognostic biomarkers from metabolism-related genes.
The risk scores of STAD patients in the testing and internal validation sets were further calculated using the same coefficients. Patients were sub-grouped using the same cutoff value as the training set. The corresponding ROC curve and Kaplan-Meier survival curves for the TCGA testing set and the entire TCGA dataset showed that the AUCs of the signature remained high and the high-risk groups had consistently shorter PFS periods than the low-risk groups (Figure 6).
In order to select the hub genes that may significantly modulate cancer metabolism in GC, WGCNA co-expression analysis was firstly conducted and the PPI network was constructed. A total of 20 genes that strongly correlated with the two metabolic subtypes and had the most connections within the PPI network were screened out and considered as candidates for the construction of the prognostic model. Using the Lasso regression analysis, a six-gene (DYNC1I1, GPER1, MFAP2, ARRB1, C3 and GLI1)signature was identified after the verification of the training, testing, and external validation sets which included a total of 639 GC patients from the TCGA-STAD and GSE62254 dataset. The model interpreted the information of gene expression into risk score for the accurate estimation of prognosis in GC. The results of survival analyses and time-dependent ROC analyses in each set revealed that the signature had stable performance in discriminating high-risk and low-risk GC patients. Notably, the 5-year AUCs for the signature in the whole TCGA-STAD dataset and GSE62254 dataset were 0.72 and 0.70, respectively. Furthermore, subgroup analysis confirmed that the signature performed well in risk prediction among GC patients with different clinical and pathological features. When clinicopathologic parameters were taken into consideration, the constructed risk-score system could still independently predict the prognosis of GC patients. A nomogram integrating the calculated risk score and clinical information was ultimately constructed for the accurate prediction of survival probability of GC patients. The nomogram showed confident clinical utility and outperformed the individual predictor in GC.
The survival status of the high-risk and low-risk subgroups was compared by Kaplan-Meier (K-M) survival analysis. Time-dependent Receiver operating characteristic(ROC) curve analysis was conducted to assess the prognostic value of the identified model using the R package timeROC[24]. The independence of the prognostic signature in the survival prediction of STAD patients was evaluated using univariate and multivariate Cox regression analyses. The prognostic performance of the signature was similarly evaluated in the TCGA testing and GEO external validation set. The immune status of tumor samples such as immune cells infiltration and tumor purity was compared between different subtypes in the TCGA-STAD dataset using Wilcoxon test. All statistical analyses were using R 3.6.0 (https://mirrors.tuna.tsinghua.edu.cn/CRAN/) with default software parameters.value < 0.05 was considered significant statistically.
Several previous studies have also identified specific prognostic models for the risk prediction of GC. For example, Lv[50] proposed a seven-gene signature which contained TGFB1, EGF, MKI67, ILF3, INCENP, TNPO2 and CHAF1A. Jiang[51]identified a biomarker consisting of 16 immune-related genes such as HSPA1A,HSPA1B, HSPA5. Yang[28] discovered another immune-related signature containing 10 genes such as NRP1 and TNFRSF18 that was totally different from that of Jiang[51]. The prognostic performance of the present model was further compared with that of the three previous models. Among the four different signatures,this six-gene biomarker had the highest C-index and AUC values. It could be concluded that this energy metabolism-related signature outperforms some previous biomarkers in the survival prediction of GC patients, and has great potential to be used for clinical application in the future.
However, there are still some limitations of this study. For example, the analysis was based on just retrospective data and needs to be verified in a prospective cohort containing samples from multi-centers before clinical application. Deeper mechanism research was also needed to elucidate the exact functions of the identified signature in GC.
CONCLUSION
In summary, by analyzing the expression levels of energy metabolism-related genes in GC tumor tissues, two different clusters with varied clinical characteristics, clinical outcomes, and immune status were identified in the TCGA-STAD dataset. A prognostic signature containing six metabolism-related genes and a novel nomogram was identified for the accurate risk prediction of GC patients.
ARTICLE HIGHLIGHTS
Research background
Energy metabolism has always been a hallmark of cancer cells and the complex metabolic characteristics of tumor cells can greatly influence the clinical fate of malignancies.
Research motivation
A deep understanding of the cancer metabolic fingerprint may be crucial to the development of new therapies and in identifying promising prognostic signatures.
Research objectives
To select key prognostic factors of gastric cancer (GC) among the 587 energy metabolism genes, and construct a potential metabolism-related model for the survival prediction of GC patients.
Research methods
We trained and verified the energy metabolism-related gene signature among a total of 339 GC samples from The Cancer Genome Atlas (TCGA) Stomach Adenocarcinoma STAD) dataset and 300 tumor samples from the GSE62254 dataset of the Gene Expression Omnibus.
Research results
We successfully created a prognostic model based on energy-metabolism-related gene expression profiles in primary stomach adenocarcinoma based on an analysis of the TCGA-STAD and GSE62254 datasets. We were able to divide and identify different subtypes for prognosis and develop a risk score based on 6 gene signatures to potentially stratify the prognosis of individuals which was validated in a second cohort.
Research conclusions
In summary, by analyzing the expression levels of energy metabolism-related genes in GC tumor tissues, two different clusters with varied clinical characteristics, clinical outcomes, and immune status were identified in the TCGA-STAD dataset. A prognostic signature containing six metabolism-related genes and a novel nomogram was identified for the accurate risk prediction of GC patients.
Research perspectives
This study demonstrates the possibility of the risk score calculated with combination of gene expression in energy metabolism-related prognostic models.
Oh, he was excited! Now Justin had a way to know when his new baby sister would come to play. From that moment on, he checked the old plum tree several times a day and reported his findings to me. Of course, he was quite concerned in November when all the leaves fell off the tree. By January, with the cold and the rains, he was truly worried whether his baby would be cold and wet like his tree. He whispered to my tummy that the tree was strong and that she (the baby) had to be strong too, and make it through the winter.
The authors would like to thank all researchers who contributed to the The Cancer Genome Atlas and Gene Expression Omnibus data sets included. An earlier version of it has been presented as preprint in Research square according to the following link:https://www.researchsquare.com/article/rs-138125/v1.
 World Journal of Gastrointestinal Oncology2022年2期
World Journal of Gastrointestinal Oncology2022年2期
- World Journal of Gastrointestinal Oncology的其它文章
- Endoscopic ultrasound-guided ablation of solid pancreatic lesions:A systematic review of early outcomes with pooled analysis
- Prevention of late complications of endoscopic resection of colorectal lesions with a coverage agent:Current status of gastrointestinal endoscopy
- Predictive value of serum alpha-fetoprotein for tumor regression after preoperative chemotherapy for rectal cancer
- Chemotherapy predictors and a time-dependent chemotherapy effect in metastatic esophageal cancer
- Association and prognostic significance of alpha-L-fucosidase-1 and matrix metalloproteinase 9 expression in esophageal squamous cell carcinoma
- Multimodal treatment in oligometastatic gastric cancer