
Catalytic performance improvement of volatile organic compounds oxidation over MnOx and GdMnO3 composite oxides from spent lithium-ion batteries:Effect of acid treatment
2021-09-02 12:45:54MingmingGuoLizhongLiuJiananGuHongboZhangXinMinJianxingLiangJinpingJiaKanLiTonghuaSun
Mingming Guo,Lizhong Liu,Jia-nan Gu,Hongbo Zhang,Xin Min,Jianxing Liang,Jinping Jia,4,Kan Li,4,*,Tonghua Sun,5,*
1 School of Environmental Science and Engineering,Shanghai Jiao Tong University,Shanghai 200240,China
2 School of Chemistry and Chemical Engineering,Nantong University,Nantong 226019,China
3 School of Chemical and Environmental Engineering,Shanghai Institute of Technology,Shanghai 201418,China
4 Shanghai Institute of Pollution Control and Ecology Security,Shanghai 200092,China
5 Shanghai Engineering Research Center of Solid Waste Treatment and Resource Recovery,Shanghai 200240,China
Keywords: Spent lithium-ions batteries Acid treatment Multi manganese-based oxides and perovskite VOCs oxidation
ABSTRACT In this work,cathode materials of spent lithium-ion ternary batteries are recovered and used as metal precursor to prepare multi-metal oxides MnOx(SY)and GdMnO3(SY)via combustion method and sol–gel method,respectively.Furthermore,a series of MnOx(SY)-n and GdMnO3(SY)-n(n=0.05,0.10,1.00,4.00,n represents the dilute HNO3 concentration)catalysts are fabricated by acid treatment of MnOx(SY)and GdMnO3(SY)samples and catalytic activities of oxygenated VOCs oxidation over all the prepared catalysts are investigated.Catalytic evaluation results show that acid-treated MnOx(SY)-0.10 and GdMnO3(SY)-0.05 samples perform the optimum VOCs removal efficiency respectively,which may be attributed to their obvious enhancement of physicochemical properties.In detail,MnOx(SY)-0.10 and GdMnO3(SY)-0.05 samples exhibit the larger specific surface area,bigger amount of surface high-valence metal ions(Mn4+,Co3+,Ni3+),more abundant adsorbed oxygen species and better low-temperature reducibility,which can play a crucial role in the significant improvement of VOCs oxidation.In situ DRIFTS results imply that the possible main intermediates are-OCO,-COO and-C-O species produced during VOCs oxidation.Possible by-products are further determined via TD/GC–MS analysis.
1.Introduction
Volatile organic compounds(VOCs),mainly emitted from the industry and transportation,are considered as major air pollutants[1–3]and then many technologies have been investigated and applied to control VOCs emission,such as adsorption,thermal combustion,photocatalytic degradation,plasma degradation,catalytic oxidation and so on[2,4–6].Among them,catalytic oxidation has been highlighted as one of the most promising technologies [7–10].Previous studies have shown that noble metal and its supported catalysts can exhibit excellent catalytic performance of VOCs oxidation but its drawbacks such as high cost prevented its wide application [11–14].In contrast,transition metal oxides have been widely used as the high-performance catalysts for VOCs removal attributed to its low price[15–18].Manganese oxides and manganese perovskites(AMnO3)are often considered as effective candidates for VOCs removal[7,12,19].However,individual MnOxand AMnO3catalysts exhibit lower catalytic performance than those of binary or multi manganese-based oxides in VOCs deep oxidation[20–24].
In the past decades,spent lithium-ion batteries(LIBs)have attracted public attention and valuable metals such as high purity of Li and Co can be derived from spent LIBs via hydrometallurgical technology[25,26].While excessive and complicated separation and purification steps can result in lots of water and energy waste and also bring secondary pollution to the environment[27].If multi transition metals such as Mn,Co,Ni and Cu can be together recovered from spent LIBs by simple steps and then they were used as metal precursor to prepare catalysts,this method can not only realize the high-efficiency recovery of metal resources but also achieve pollutants control.Based on the previous work [27],various transition metal such as Mn,Ni,Co and Cu were recycled from spent ternary LIBs and they were used as active metal components to prepare manganese-based and manganese-based perovskite oxides catalysts for VOCs removal.
It has been reported that catalytic activities of VOCs oxidation are often affected by their properties such as specific surface area,surface morphology,high-valence metal ions,active oxygen species and lowtemperature reducibility[28–30].Recent studies have shown that acid treatment method can effectively affect the physicochemical properties of catalysts and furthermore,suitable acid concentration contributes to creating more structural defects,changing surface morphology and enhancing the generation of high-valence metal ions and active oxygen species,which has a great influence on improving VOC conversion[1,3,31,32].Herein,a series of acid-treated manganese-based oxides were synthesized from cathode materials of spent ternary LIBs and effect of dilute HNO3concentration on catalytic behavior of VOCs oxidation over prepared catalysts was investigated.In this work,typical VOCs such as toluene was selected as VOCs source and also oxygenated 2-ethoxy-ethanol is used as molecule probe because 2-ethoxy-ethanol is often used as an excellent solvent for synthetic resin and paint as well as a dispersant,but its volatility can bring about damage to human health such as mucosal irritation and headache.
2.Experimental
2.1.Metal precursor solution derived from spent lithium-ions ternary batteries
Acid leaching solution of cathode materials from spent lithium-ions ternary batteries were obtained according to the following steps[27].Firstly,cathode materials were recovered from spent lithium-ions ternary batteries by complete discharging and physical dismantling.Secondly,cathode materials were calcined at 350 °C for 30 min in a muffle furnace to remove volatile substances and then small fragments of cathode materials were completely grinded into powder.Thirdly,grinded powder was treated based on the conditions of 20 g·L?1of solid–liquid ratio,1.5 mol·L?1of citric acid,0.3 g·g?1of glucose and 80 °C of treatment temperature and then the acid leaching solution was obtained.Subsequently,the content of each element in the acid leaching solution was measured by ICP-AES,as shown in Table 1.In order to effectively remove Li and Al element,3 g oxalic acid was added into 80 ml of acid leaching solution and element content of obtained sediment was determined by ICP-AES as displayed in Table 1.Metal precursor solution was obtained by completely dissolving the sediment above into 30 ml dilute of HNO3.
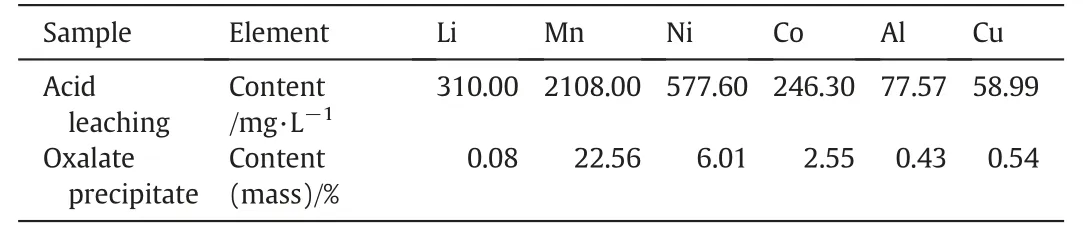
Table 1ICP-AES results of acid leaching solution and oxalate precipitate
2.2.Catalyst preparation
In this work,all the chemical reagents were analytical grade and directly used without any other purification process.Manganese-based perovskite oxides were prepared using sol–gel method[5].The specific preparation process was as follows:the usage amount of metal nitrate,Gd(NO3)3·6H2O and citric acid(C6H8O7·H2O)were determined according to the stoichiometric ratio of 1:1:1.5,respectively.Next,the weighed chemical reagents were dissolved into 50 ml of deionized water in a beaker and then the metal precursor solution was treated at 80 °C under strong stirring in a water bath until most of deionized water was evaporated.Subsequently,the viscous colloid was dried at 120°C in an oven overnight and finally the dried gel material was calcined in a muffle furnace.Firstly,the calcination temperature increased up to 200°C kept for 2 h and then it continued to increase to 750°C maintained for 3 h and then the calcined powder was grinded to obtain GdMnO3(SY)sample.
Manganese-based oxides were prepared by a traditional combustion method[32]using metal nitrate from cathode materials as the metal precursor.Metal precursor solution was firstly dissolved into 50 ml of deionized water and 1.5 times stoichiometry of citric acid was added into the metal solution drop by drop under the magnetic stirring.The metal mixture solution was treated at 80°C in a water bath until most water was lost and then placed in an oven kept 120 °C overnight.At last,the dried material was calcined at 400°C for 3 h in a muffle furnace and the obtained sample was denoted as MnOx(SY).To make a comparison,pure GdMnO3and MnOxwere prepared according to the same synthesis process.
Acid-treated GdMnO3(SY) and MnOx(SY) samples with different concentrations of dilute HNO3were obtained according to the followed steps.In detail,each sample of GdMnO3(SY)and MnOx(SY)were immersed into 50 ml of dilute HNO3solution with 0.05 mol·L?1,0.10 mol·L?1,1.00 mol·L?1,4.00 mol·L?1respectively and then each mixture solution was treated under vigorous stirring at room temperature for 0.5 h and followed by filtering operation.Secondly,the filter residues were washed by deionized water and collected,dried at 120°C in an oven overnight.Finally,the obtained acid-treated samples were denoted as GdMnO3(SY)-n,MnOx(SY)-n(n=0.05,0.10,1.00 and 4.00,n represents the concentration of dilute HNO3)catalysts,respectively.
2.3.Catalyst characterization
X-ray diffraction was measured using a Lab X-XRD-6100 diffractometer(Shimadzu,Japan)(CuKαray,scanning angle 5°–85°).N2adsorption–desorption isotherms were completed on the Autosorb-iQ analyzer of Quantachrome Corporation in America to determine the specific surface area and pore structure parameters of prepared catalysts.The sample was firstly degassed at 300°C for 6 h under vacuum and after that,N2adsorption–desorption measurement was performed at ?196°C.The specific surface area,pore volume and pore diameter of the sample was calculated using the BET and BJH method,respectively.SEM images were observed with the JEOL JSM-7800F scanning electron microscope(Japan)for the surface morphology of the samples.Element composition and valence states on the surface of the sample were analyzed using a Thermo Scientific iCAP6300 Series X-ray photoelectron spectroscopy (XPS) instrument and AlKαwas used as the radiation source and the binding energy was corrected based on C1s(284.8 eV).H2temperature-programmed reduction(H2-TPR)of the sample was measured on a FINESORB-3010D instrument of FINETEC in China.The sample was placed into a U-shaped quartz tube and the temperature was firstly increased to 300 °C for 1 h in Ar gas (15 ml·min?1) and then cooled down to room temperature.Subsequently,the sample was immersed into a mixture gas of 10% H2/Ar mixture (15 ml·min?1)and then increased up to 900 °C from room temperature at a heating rate of 10°C·min?1.The intermediates species during EE oxidation were detected by in-situ DRIFTS experiment on a Nicolet 6700 Fourier transform infrared spectrometer from Thermo Scientific in American.In addition,in-situ cell with KBr window was selected and it was equipped with a liquid N2-cooled MCT detector.Before each experiment,the sample in situ pool was pretreated at 300°C for 60 min in air or N2with a total flow of 50 ml·min?1to remove gas impurities.The background spectra in the range of 400–4000 cm?1were collected and the spectral resolution and scan times are 4 cm?1and 100,respectively.Possible by-products produced during EE oxidation were further investigated on an Agilent/7890B-5977B Thermal Desorption/Gas Chromatograph Mass Spectrometer(TD/GC–MS)instrument(American).
2.4.Catalytic evaluation
Catalytic activities of VOCs oxidation over prepared catalysts were measured in a fixed quartz-tube reactor with an inner diameter of 6 mm and a length of 40 cm.50 mg of catalyst powder was placed in a quartz tube and fixed using some amount of quartz wool.Firstly,the quartz tube loaded with the sample was placed in the middle of electronic tube furnace with a thermocouple to detect the reaction temperature.Secondly,the sample was pre-treated at 340°C for 60 min under the air flow of 50 ml·min?1to remove other gas purities adsorbed on the sample.Thirdly,the inlet reaction gas was switched into 1000 ppm VOCs mixture at the same total flow of 50 ml·min?1using artificial air as balance gas with the corresponding GHSV of 60,000 ml·g?1·h?1and VOCs was detected on HXSP GC-950 gas chromatography.In addition,the gas flow rate was controlled using mass flowmeter and EE gas was blown out by constant bubbling from liquid VOCs placed in an ice-water bath.VOCs conversion and CO2yield were calculated by the following Eqs.(1)and(2),in which n stands for the number of VOCs molecule.

3.Results and Discussion
3.1.Crystal structure
The crystal structure of prepared samples is investigated by powder X-ray diffraction and the XRD results are shown in Fig.1.As depicted in Fig.1(A),XRD peaks of GdMnO3,GdMnO3(SY),GdMnO3(SY)-0.05,GdMnO3(SY)-0.10,GdMnO3(SY)-1.00 and GdMnO3(SY)-4.00 catalysts are dominant by GdMnO3(PDF # 25-0337) and are co-existed with other diffraction peaks at 15.56°,28.76°,35.45°,38.03° and 31.68°,35.83° in accordance with those of GdMn2O5(PDF # 52-0301) and Gd2O3(PDF#43-1015),respectively.After acid treatment,it can be found that the intensity of crystalline structure located at 28.76°(GdMn2O5) gradually grows higher while the crystallinity degree at 33.15°becomes weaker,which may be owing that the presence of dilute acid can induce the Mn3+ions to convert into Mn4+and Mn2+and then some amount of Mn3+ions in GdMnO3were substituted by Mn4+[5].Meanwhile,partial of solid Gd3+can be selectively removed.This result reveals that most intensive peaks of GdMnO3catalyst can be obviously affected by dilute HNO3and the amount of Mn4+ions was obviously enhanced.In the case of MnOx,its main characteristic peaks can be assigned to typical Mn3O4(PDF#24-0734)and meanwhile some XRD peaks related to Mn2O3(PDF # 41-1442) were visible at 32.95° and 40.62°.In terms of MnOx(SY),MnOx(SY)-0.10,MnOx(SY)-0.10,MnOx(SY)-1.00 and MnOx(SY)-4.00 catalysts,the major crystal compositions are Cu1.5Mn1.5O4(PDF#35-1172).Compared with other catalysts,two obvious peaks at 33.03°and 55.30°can be respectively observed over MnOx(SY)-0.10 catalyst,which are assignable to CoMn2O4(PDF#01-1126)and Co2NiO4(PDF#02-1074),indicating that appropriate acid concentration contributes to the formation of crystal phase.
3.2.Specific surface area and pore size distribution
Specific surface area and pore size distribution of prepared samples were determined via N2adsorption and desorption isotherms as shown in Fig.2 and the corresponding values of SBETand average pore diameter(d)are listed in Table 2.From Table 2,it can be seen that the specific surface area showed an significant increased trend from single metal oxide MnOx(11.519 m2·g?1),GdMnO3(15.379 m2·g?1) to multi metal oxides MnOx(SY) (185.892 m2·g?1),GdMnO3(SY)(20.458 m2·g?1),corresponding that the average pore diameter decreased from MnOx(17.411 nm),GdMnO3(30.795 nm)to multi metal oxides MnOx(SY)(6.651 nm),GdMnO3(SY)(3.054 nm),respectively.This increase of specific surface area may be related to the decrease of average pore diameter,which may be caused during the calcined process of multi metal components which can be confirmed by EDS as shown in Table 2.After acid treatment,by comparison,acid-treated MnOx(SY)-0.10 and GdMnO3(SY)-0.05 can exhibit the largest specific surface area of 189.112 m2·g?1,27.461 m2·g?1,respectively.No obvious change of average pore diameters can be observed between MnOx(SY)-0.10,GdMnO3(SY)-0.05 and MnOx(SY),GdMnO3(SY) samples.The increased specific surface area contributed to providing more active sites which are beneficial to the adsorption and diffusion of VOC molecule,thus enhancing the catalytic activity of VOC oxidation[32].
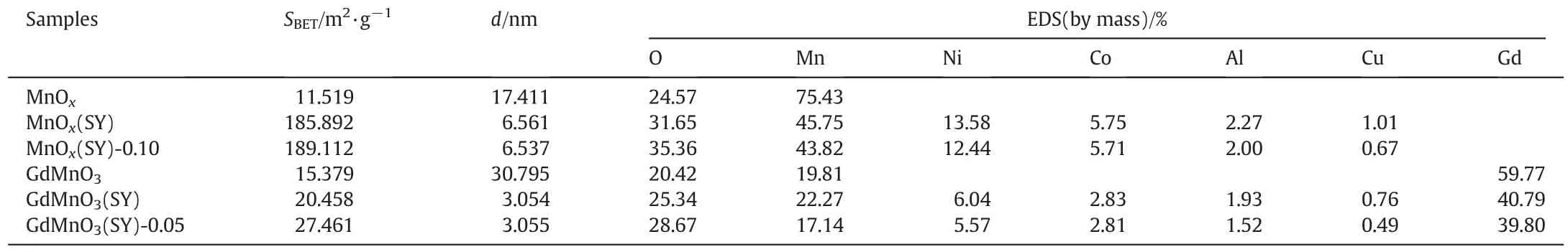
Table 2Specific surface area,average pore diameter and EDS elements content
3.3.Surface morphology
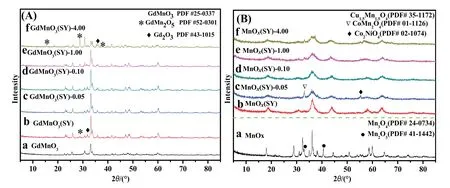
Fig.1.XRD patterns of(A)a-GdMnO3,b-GdMnO3(SY),c-GdMnO3(SY)-0.05,d-GdMnO3(SY)-0.10,e-GdMnO3(SY)-1.00,f-GdMnO3(SY)-4.00 and(B)a-MnOx,b-MnOx(SY),c-MnOx(SY)-0.05,d-MnOx(SY)-0.10,e-MnOx(SY)-1.00,f-MnOx(SY)-4.00.
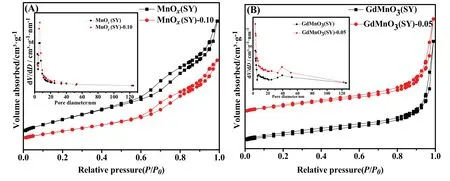
Fig.2.N2 adsorption–desorption isotherms and pore size distribution of(A)MnOx(SY),MnOx(SY)-0.10 and(B)GdMnO3(SY),GdMnO3(SY)-0.05.

Fig.3.SEM images of MnOx(a,b),MnOx(SY)(c,d),MnOx(SY)-0.10(e,f),GdMnO3(g,h),GdMnO3(SY)(i,j),GdMnO3(SY)-0.05(k,l).
Fig.3 reveals the SEM images of surface morphologies and structural features over prepared samples.In terms of manganese-based oxides,the surface morphology of MnOx(a,b)catalyst displayed disordered entities with aggregated macroblocks which may restrict the specific surface area.Compared with MnOx(a,b),MnOx(SY)(c,d) sample presented a smoothly layered structure with numerous smaller interspace and lots of nanopores which can lead to the increase of specific surface area.And then,after acid treatment,many thin flakes were visible and meanwhile a number of irregular particles were formed on the surface of MnOx(SY)-0.10(e,f) catalyst.In terms of manganesebased perovskite oxides,initial GdMnO3(g,h)presented an irregularly aggregated granular and block entities while GdMnO3(SY)sample exhibited a smooth surface with numerous microporous gaps and additionally the obviously smooth surface can be observed after acid treatment with low HNO3concentration which may result in the enhanced specific surface area.Therefore,it was confirmed that acid treatment can increase the specific surface area by modifying the surface morphologies and porous structure to some extent,which can facilitate the oxidation reaction of VOC molecule[3].
3.4.Oxygen species and metal valence
XPS experiments were conducted to determine the surface element composition,metal oxidation states and oxygen species on the surface of prepared catalysts.The corresponding O1s,Mn2p3/2,Co2p3/2 and Ni2p3/2 XPS spectra on the surface of MnOx,MnOx(SY),MnOx(SY)-0.10,GdMnO3,GdMnO3(SY) and GdMnO3(SY)-0.05 catalysts were displayed in Fig.4.In addition,the surface molar ratio of Oads/Olatt,Mn4+/Mn3+,Mn3+/Mn2+,Co3+/Co2+and Ni3+/Ni2+were calculated from corresponding areas according to curve-fitting method and then they were summarized at Table 3.The O1s peaks of prepared catalysts were shown in Fig.4(A,B)and two forms of oxygen species can be obtained: surface lattice oxygen (Olatt) located at 529.3–529.6 eV and adsorbed oxygen (Oads) viewed at 531.1–531.5 eV [5,12].It can be clearly seen from Table 3 that the sequence of Oads/Olattratio from the highest to the lowest value was MnOx(SY)-0.10(1.62)>MnOx(SY)(0.98) > MnOx(0.41) for manganese-based oxides catalysts and GdMnO3(SY)-0.05(1.00)>GdMnO3(SY)(0.73)>GdMnO3(0.70)for manganese-based perovskite catalysts,respectively,suggesting that the catalysts prepared from spent lithium-ions batteries possessed much abundancy oxygen vacancies compared with pure MnOx and GdMnO3.Furthermore,the oxygen vacancies of MnOx(SY) and GdMnO3(SY)samples can be enriched by using certain concentration of HNO3.As shown in Fig.4(C,D),the asymmetrical Mn2p3/2 spectrum of individual catalyst could be decomposed into three peaks at high binding energies of 643.9–644.7 eV,at middle binding energies of 641.8–642.6 eV,at low binding energies of 640.5–641.3 eV,which were indicative of the surface Mn4+,Mn3+,Mn2+species,respectively[5,12].From Table 3,it can be found that the value of surface Mn4+/Mn3+increased from 0.55 of MnOxto 0.72 of MnOx(SY)-0.10,while the Mn3+/Mn2+ratio remarkably decreased from 1.84 of MnOxto 0.96 of MnOx(SY)-0.10.By comparison,it can be obtained that there is relatively higher value of Mn4+/Mn3+ratio over GdMnO3(SY)and GdMnO3(SY)-0.05 samples.It should be noted that the Mn4+/Mn3+ratio of GdMnO3(SY) (0.77) is slightly higher than that (0.73) of GdMnO3(SY)-0.05 while GdMnO3(SY)-0.05 exhibits greater value(2.75)of Mn3+/Mn2+than that(1.43)of GdMnO3(SY),indicating thatpartial Mn4+will be converted into Mn3+under the condition of acid treatment due to the co-existence of Mn4+and Ni2+,Co2+,which were consistent with the higher ratio of Co3+/Co2+and Ni3+/Ni2+as shown in Table 3.This result implied that there is strong electrons transfer between Mn4+and Ni2+,Co2+over GdMnO3(SY)-0.05 catalyst immersed into the dilute HNO3solution.It should be noted that,the Co2p3/2 XPS peaks of each catalyst displayed two components located at 779.8–780.2 eV due to the presence of surface Co3+,and at 781.4–782.5 eV corresponding to the surface Co2+,as well as the existence of the satellite peak at 786.0–786.6 eV which can be used to confirm the presence of Co2+[27]as shown in Fig.4(E).And meanwhile,as displayed in Fig.4(F),Ni2p3/2 spectra exhibits three peaks at 855.9–856.6 eV assigned to surface Ni3+,at 854.5–854.8 eV attributed to Ni2+-and at 861.2 eV ascribed to shake up satellite[27].Therefore,acid treatment has an obvious influence on accelerating the disproportionation of Mn3+to Mn4+and Mn2+,accompanying with the abundant generation of oxygen vacancies which is beneficial to the adsorption,diffusion and generation of active oxygen species.It is well-known that the enhanced amount of high-valence metal ions(Mn4+,Mn3+,Ni3+Co3+species)can result in the oxidation activity of VOC molecule.
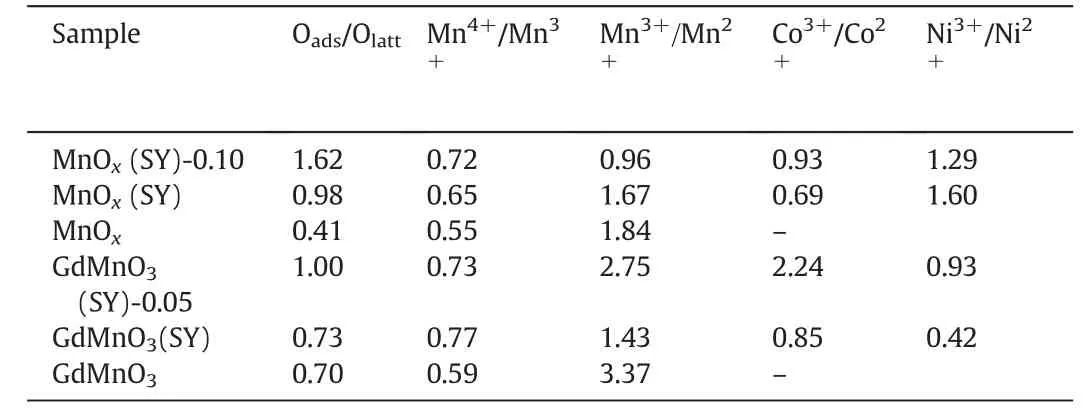
Table 3Surface elements component and their oxidation states
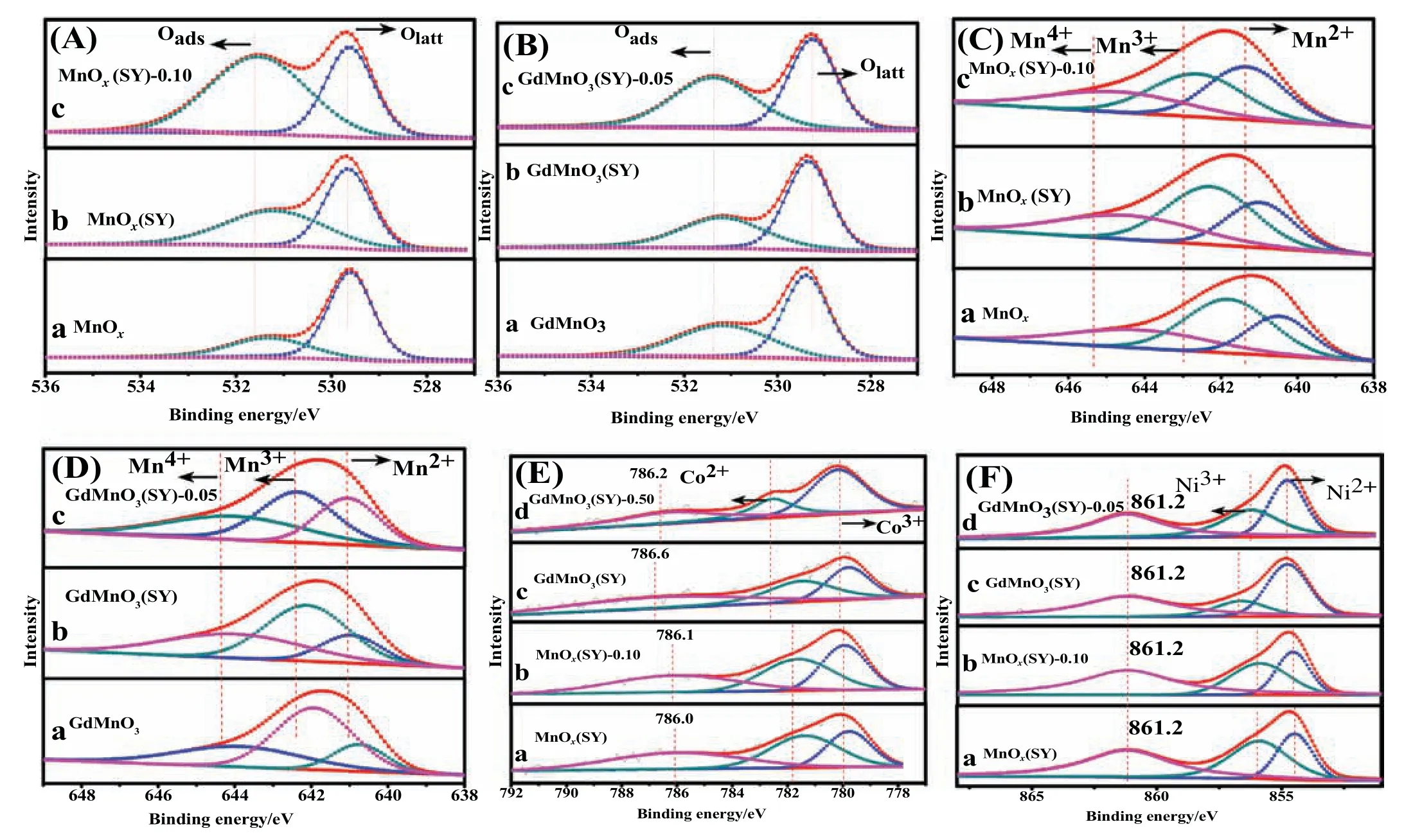
Fig.4.XPS spectra of(A)O1s:a-MnOx,b-MnOx(SY),c-MnOx(SY)-0.10 and(B)O1s:a-GdMnO3,b-GdMnO3(SY),c-GdMnO3(SY)-0.05;(C)Mn2p3/2:a-MnOx,b-MnOx(SY),c-MnOx(SY)-0.10 and(D)Mn2p3/2:a-GdMnO3,b-GdMnO3(SY),c-GdMnO3(SY)-0.05;(E)Co2p3/2:a-MnOx(SY),b-MnOx(SY)-0.10,c-GdMnO3(SY),d-GdMnO3(SY)-0.05;(F)Ni2p3/2:a-MnOx(SY),b-MnOx(SY)-0.10,c-GdMnO3(SY),d-GdMnO3(SY)-0.05.
3.5.H2 Temperature-programmed reduction
The reduction behavior of prepared catalysts was examined by H2-TPR experiments,and TPR profiles of MnOx(a),MnOx(SY)(b),MnOx(SY)-0.10 (c),GdMnO3(d),GdMnO3(SY) (e) and GdMnO3(SY)-0.05(f)were depicted in Fig.5(A)and(B).For MnOxcatalyst,the reduction peaks centered at 317 and 451°C may associated with the removal of adsorbed oxygen species as well as the reduction of Mn4+to Mn3+,the reduction of Mn2O3to Mn3O4coexisting with the reduction of partial Mn3O4to MnO,respectively[5,12,28,29].In contrast to pure MnOx,TPR profiles of MnOx(SY) and MnOx(SY)-0.10 can be separated into three reduction peaks,in which the first peaks centered at 246 and 241°C,the second peaks located at 328 and 322°C and the last peaks maximum at 470 and 466 °C may be assigned to the desorption of adsorbed oxygen species occurring with the reduction process of MnO2to Mn2O3,the reduction step of Mn2O3to Mn3O4,Mn2O3to Mn3O4,respectively [19].By comparison,it can be clearly seen that MnOx(SY) and MnOx(SY)-0.10 exhibited better low-temperature reducibility than that of pure MnOx,while the TPR peaks of MnOx(SY)-0.10 slightly shifted to low temperature compared with MnOx(SY),suggesting that the acid etching of 0.10 mol·L?1HNO3has a little influence on facilitating the reduction performance of MnOx(SY).As shown in Fig.5(B),three were two obvious reduction peaks centered at 450,668 °C and a weak reduction peak located at 730 °C for GdMnO3,corresponding to the reduction steps of Mn2O3to Mn3O4as well as Mn3O4to MnO,the reduction process of Mn3O4left to MnO[33,34],and the reduction of lattice oxygen in the bulk of GdMnO3,respectively.In the case of GdMnO3(SY)catalyst,it possessed a shoulder reduction peak about at 325°C which may be ascribed to the reduction process of MnO2→Mn2O3→Mn3O4→MnO,and a high temperature reduction peak viewed at 614°C which can be attributed to Mn3+left in the bulk to Mn2+.With respect to GdMnO3(SY)-0.05 catalyst,the reduction peak of MnO2→Mn2O3→Mn3O4shifted obviously to lower temperature at 298 °C than that of GdMnO3(SY).In addition,the latter two reduction peaks placed at 624 and 728 °C may be due to the Mn3+left to Mn2+and the reduction of lattice oxygen in the bulk,respectively.On the contrary,GdMnO3exhibited the worst reducibility at low temperature while GdMnO3(SY)-0.05 catalyst performed the best reducibility performance below 500 °C,indicating that the acid etching of 0.05 M HNO3could enhance the low-temperature redox ability of GdMnO3(SY)-0.05 catalyst.It is generally accepted that the enhancement of low-temperature reducibility of the sample can give rise to the improvement of catalytic activity of VOC oxidation,implying that acid treatment could lead to better catalytic behavior in the VOC oxidation.
3.6.Catalytic activity in VOCs oxidation

Fig.5.H2-TPR profiles of(A):a-MnOx,b-MnOx(SY)and c-MnOx(SY)-0.10;and(B):d-GdMnO3,e-GdMnO3(SY)and f-GdMnO3(SY)-0.05.

Fig.6.2-ethoxy-ethanol conversion and CO2 yield over(A,a):MnOx,MnOx(SY),MnOx(SY)-0.10,MnOx(SY)-0.10,MnOx(SY)-1.00,MnOx(SY)-4.00;(B,b):GdMnO3,GdMnO3(SY),GdMnO3(SY)-0.05,GdMnO3(SY)-0.10,GdMnO3(SY)-1.00,GdMnO3(SY)-4.00.
The catalytic activities of 2-ethoxy-ethanol oxidation over prepared catalysts were evaluated in the reaction temperature range of 140–340 °C and the results were plotted in Fig.6.The values of T50%and T90%,corresponding to the reaction temperatures for 50%and 90%2-ethoxy-ethanol conversion,respectively,were used to compare the catalytic performance.As can be seen from Fig.6(A),MnOxdisplayed the worst performance of 2-ethoxy-ethanol oxidation whose values of T50%and T90%were 186 and 285 °C,respectively.In comparison with MnOx,a significant increase can be observed over MnOx(SY),MnOx(SY)-0.10,MnOx(SY)-0.10,MnOx(SY)-1.00 and MnOx(SY)-4.00 catalysts.Among the five manganese-based oxides,MnOx(SY)-0.10 catalyst presented an outstanding removal efficiency for 2-ethoxy-ethanol conversion with the T50%and T90%of 145 and 176°C,respectively.The disparity of about 16°C and 42°C can be observed between MnOx(SY)and MnOx(SY)-0.10 catalysts at 50%and 90%of 2-ethoxy-ethanol conversion,respectively.In addition,some obvious difference can be found between MnOx(SY)-0.05(T50%=150 and T90%=203°C),MnOx(SY)-1.00(T50%=167 and T90%=234°C)and MnOx(SY)-4.00(T50%=180 and T90%=270 °C) catalysts.As for manganese-based perovskite oxides,the six catalysts in this study can be ranked in the decrease order in terms of degradation efficiency of 2-ethoxy-ethanol oxidation:GdMnO3(SY)-0.05 (T50%=189 and T90%=224 °C) > GdMnO3(SY)-0.10 (T50%=193 and T90%=233 °C) > GdMnO3(SY) (T50%=201 and T90%=240 °C) > GdMnO3(T50%=209 and T90%=250 °C) >GdMnO3(SY)-1.00(T50%=215 and T90%=251°C)>GdMnO3(SY)-4.00 (T50%=220 and T90%=258 °C).Obviously,the values of T50%and T90%over the GdMnO3(SY)-0.05 catalysts were 12 and 16 °C lower than those over the GdMnO3(SY) catalyst,respectively.Meanwhile,the T50%and T90%values of GdMnO3are 8 and 10°C higher in comparison to those of GdMnO3(SY).As a consequence,removal efficiency of 2-ethoxy-ethanol deep oxidation over manganese-based oxides or perovskite oxides can be significantly affected by acid etching using appropriate dilute HNO3.In addition,catalytic activities of toluene oxidation over MnOx(SY),MnOx(SY)-0.10 and GdMnO3(SY),GdMnO3(SY)-0.05 catalysts were evaluated,as shown in Fig.7.It can be seen that MnOx(SY)-0.10(T50%=220°C and T90%=245°C)and GdMnO3(SY)-0.05(T50%=234°C and T90%=256°C)performed better performance than those of MnOx(SY)(T50%=240°C and T90%=260°C)and GdMnO3(SY)(T50%=244°C and T90%=276°C).And meanwhile,compared with manganese oxides [38–41]in previous works as listed in Table 4,acid-treated MnOx(SY)-0.10 and GdMnO3(SY)-0.05 exhibited much excellent catalytic abilities.It has been revealed that there was an intrinsic relation between catalytic activity of the catalyst and its specific surface area,surface pore structure,reductive ability,active metal species oxygen species.In this work,acid-treated MnOx(SY)-0.10 and GdMnO3(SY)-0.05 samples presented abundant mesoporous structure,bigger specific surface areas,plenty of high-valence metal species and surface active oxygen species,better low-temperature reducibility.Therefore,it can be proved that acid treatment is an effective method to enhance the catalytic behavior of the sample.

Table 4Comparison results of VOCs oxidation over synthesized catalysts in this work and VOCs catalytic performance over manganese-based oxides in the literature
3.7.In-situ DRIFT experiment
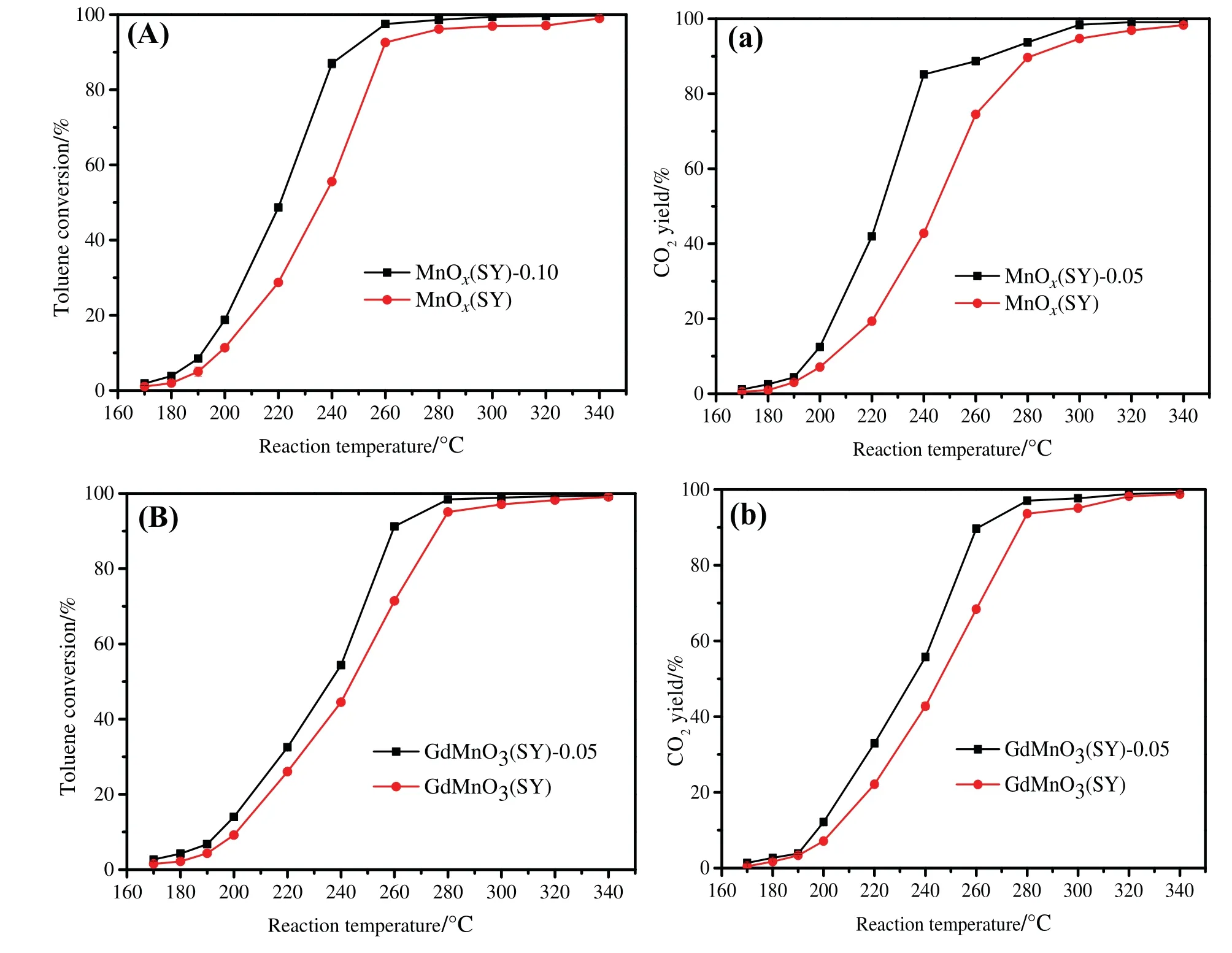
Fig.7.Toluene conversion and CO2 yield over(A,a):MnOx(SY),MnOx(SY)-0.10 and(B,b):GdMnO3(SY),GdMnO3(SY)-0.05.
Intermediate groups were firstly investigated in N2atmosphere After introducing 2-ethoxy-ethanol/N2mixture gas into reaction flow,in the case of GdMnO3(SY)-0.05 catalyst,several bands at 1630,1550,1448,1122 and 1060 cm?1can be detected as shown in Fig.8(A),which can be attributed to asymmetric stretching vibration of νas(OCO),νas(COO),symmetric C–O stretching vibration,C-O stretching modes,respectively.While no obvious difference can be observed about their intensities with reaction temperature increasing.For sample MnOx(SY)-0.10,its spectra was dominated by these bands located at 2980,2870,1600,1450,1323,1120 and 1080 cm?1.These bands at 2980,2870,1323,1120 and 1080 cm?1as displayed in Fig.8(B),corresponding to asymmetric stretching vibration νas(CH3) and νs(CH3),asymmetric stretching vibration of νas(COO) and C-O stretching modes,respectively,completely vanished as the temperature increasing to 280 °C [35,36].Whereas those bands at 1600 and 1450 cm?1could be associated with νas(OCO) and symmetric C-O stretching vibration [37],whose intensities gradually decreased along with the increasing of reaction temperature.Furthermore,in order to investigate the role of gas-phase O2in contributing to EE deep oxidation,the synthetic air(21%O2,N2as balance gas)was introduced into reaction system.In terms of GdMnO3(SY)-0.05 catalyst as shown in Fig.8(a),the relative intensities of these broad bands formed at 1580,1438 and 1378 cm?1showed a significant increase at the reaction temperature 50–150°C compared with those in N2atmosphere.Meanwhile,it could be found that all the bands intensities showed a remarkable decrease trend and all the peaks completely vanished after 280°C,indicating that the presence of gas-phase oxygen has a significant effect on the further oxidation of 2-ethoxy-ethanol which well agreed with the result of 2-ethoxy-ethanol conversion.Similarly,in the presence of O2,their peaks intensities of MnOx(SY)-0.10 gradually decreased with reaction temperature going up and they disappeared after 260 °C.It can be found that gas-phase O2molecules play a crucial role in the VOC total oxidation,which may be attributed that O2molecules can be rapidly adsorbed into oxygen vacancies and then active oxygen species can be formed and activated to facilitate the further oxidation of intermediates.

Fig.8.In-situ DRIFTS experiment during EE oxidation over GdMnO3 and MnOx under N2(99.999%)atmosphere(A,B)and air atmosphere(21%O2 and N2)(a,b)(The set reaction temperature was in the range of 50°C to 300°C).
3.8.Thermal desorption/gas chromatograph mass spectrometer
From Fig.6,it can be observed that CO2yield were much lower compared to corresponding 2-ethoxy-ethanol conversion at lower reaction temperature.It can be concluded that some by-products can be possibly produced during 2-ethoxy-ethanol partial oxidation at lower reaction temperature.Therefore,TD/GC–MS experiment was carried out to further determine the possible by-products and the TD/GS-MS analysis results were presented in Fig.9.By-products of MnOx(SY)and MnOx(SY)-0.10 catalysts were collected at 160°C as shown in Fig.9(A,a)and byproducts of GdMnO3(SY) and GdMnO3(SY)-0.05 catalysts were collected at 200°C as displayed in Fig.9(B,b).It should be noted that the by-products were firstly enriched in a Tenax TA adsorption tube.In combination with in-situ DRIFTS results,the possible by-products can be deduced as follows:ethanol;ethanol,2-ethoxy-;carbonic acid,2-ethoxyethyl 2-methoxyethyl ester; 2-propanol,1-(2-methoxy-1-methylethoxy)-; 2-ethoxyethyl acetate; acetic acid,hydroxy-,ethyl ester;acetic acid.Therefore,it can be proved that the process of EE complete conversion into CO2required a much higher reaction temperature,at which the intermediates can be further oxidized into CO2.
4.Conclusions
Multi oxides MnOx(SY)and GdMnO3(SY)are successfully prepared using cathode materials from spent ternary LIBs.Furthermore,acidtreated MnOx(SY)-n and GdMnO3(SY)-n(n=0.05,0.10,1.00,4.00)were obtained and their catalytic performance are enhanced compared with original MnOx(SY)and GdMnO3(SY).This proves that active metal from cathode materials in spent ternary LIBs can be used as metal precursor to prepare high-activity catalysts for VOCs oxidation.
MnOx(SY)-0.10 and GdMnO3(SY)-0.05 samples possess higher specific surface area,abundant pore structure,abundant high-valence metal ions and surface active oxygen species,better low-temperature reducibility.In situ DRIFTS results imply that the possible main intermediates are νas(OCO),νas(COO),-C-O species produced during the VOC oxidation and corresponding possible by-products are determined via TD/GC–MS analysis.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.

Fig.9.Possible by-products produced during EE oxidation over(A)MnOx(SY),(a)MnOx(SY)-0.10,(B)GdMnO3(SY),(b)GdMnO3(SY)-0.05.
Acknowledgements
This work was supported by the National Natural Science Foundation of China(Grant numbers 21876107 and 21607103).
 Chinese Journal of Chemical Engineering2021年6期
Chinese Journal of Chemical Engineering2021年6期
- Chinese Journal of Chemical Engineering的其它文章
- Effects of coagulation-bath conditions on polyphenylsulfone ultrafiltration membranes
- Functional monodisperse microspheres fabricated by solvothermal precipitation co-polymerization
- Synthesized graphene oxide and fumed aerosil 380 dispersion stability and characterization with partially hydrolyzed polyacrylamide
- Synthesis and characterization of caprolactone based polyurethane with degradable and antifouling performance
- Removal of lead (Pb(II)) and zinc (Zn(II)) from aqueous solution using coal fly ash (CFA) as a dual-sites adsorbent
- Application of fracturing technology to increase gas production in low-permeability hydrate reservoir:A numerical study