
Contribution of C3G and other GEFs to liver cancer development and progression
2021-05-11 09:04:52AlmudenaPorrasCeliaSequeraPalomaBragadoAlvaroGutierrezUzquizaCarmenGuerrero
Hepatoma Research 2021年5期
Almudena Porras, Celia Sequera,3, Paloma Bragado, Alvaro Gutierrez-Uzquiza, Carmen Guerrero
1Departamento de Bioquímica y Biología Molecular, Facultad de Farmacia, Universidad Complutense de Madrid, Madrid 28040,Spain.
2Instituto de Investigación Sanitaria del Hospital Clínico San Carlos (IdISSC), Madrid 28040, Spain.
3Aix-Marseille Univ, CNRS, Developmental Biology Institute of Marseille (IBDM), UMR7288, Parc Scientifique de Luminy,Marseille 13009, France.
4Instituto de Biología Molecular y Celular del Cáncer (IMBCC), Universidad de Salamanca-CSIC, Salamanca 37007, Spain.
5Instituto de Investigación Biomédica de Salamanca (IBSAL), Salamanca 37007, Spain.
6Departamento de Medicina, Universidad de Salamanca, Salamanca 37007, Spain.
Abstract Primary liver cancers constitute the fourth leading cause of cancer mortality worldwide, due to their high morbidity,late diagnosis and lack of effective treatments. Hepatocellular carcinoma (HCC) represents 80% and cholangiocarcinoma (CCA) 15% of liver cancers. Several genetic and epigenetic gene alterations (e.g., TERT, TP53 or CTNNB1) are HCC drivers, although many additional gene alterations contribute to HCC initiation and/or progression. Rho and Ras GTPases have been widely implicated in tumorigenesis and their activators (GEFs) have recently emerged as putative key players in liver cancer. The Ras GEF, C3G (RAPGEF1), a GEF mainly for Rap proteins, has recently been uncovered as a relevant gene in HCC. Its upregulation promotes tumor growth,although a decrease in C3G levels favors migration/invasion and lung metastasis. Rap1A/1B/2A/2B are overexpressed in HCC tumors, but their effects are controversial and not equivalent to those of C3G. The C3G partner, CRKL, is also overexpressed in HCC, promoting proliferation, migration and invasion. Various Rho GEFs are also deregulated in liver cancer. Tiam1 and Tiam2 expression is upregulated in HCC, promoting proliferation,migration and metastasis. In addition, ARHGEF-10L/9/19/39 are overexpressed in HCC tumors, facilitating migration, invasion, metastasis and proliferation. Another Rho GEF, Vav2, is also involved in metastasis. Little is known about the participation of these GEFs and GTPases in CCA. However, analysis of cancer databases uncovered deregulations or genetic alterations in several of these genes, in both CCA and HCC. Hence, GEFs function appear essential for liver homeostasis, although future studies are needed to define their precise function in liver cancer.
Keywords: Liver cancer, C3G, RAPGEF1, Rap, Ras GEFs, Rho GEFs, CRK, hepatocarcinoma, cholangiocarcinoma
INTRODUCTION
Primary liver cancer is the 6th most frequently diagnosed cancer and the 4th leading cause of cancer mortality worldwide[1]. It is refractory to most treatments, so life expectancy of patients is very low, with a 5-year median survival of 18%[2].
The most common type of primary liver cancer is hepatocellular carcinoma (HCC), followed by cholangiocarcinoma (CCA), a highly heterogeneous group of malignancies generated in the biliary tree,which represents approximately 15% of primary liver tumors[3]. Some classifications also include hepatoblastoma and combined HCC and CCA[4]. All these primary liver cancers can potentially originate by hepatocytes, cholangiocytes, hepatoblasts and/or liver progenitor cells. However, the cell of origin is still controversial[4]. The combined effects of oncogenic driver genes (intrinsic factors) and tumor microenvironment (extrinsic factors) determine the cancer phenotype of hepatocyte-derived liver tumors.
HCC usually occurs in the context of chronic liver disease, originated by risk factors such as infection with hepatitis B (HBV) or C viruses, alcohol abuse and nonalcoholic hepatic steatosis[2]. HCC has high molecular heterogeneity at different levels: interpatient, intertumoral (in different nodules) and intratumoral (in the same nodule).
The most frequent alterations in HCC include mutations in the TERT promoter leading toTERTreactivation;TP53alterations;CTNNB1mutations; copy number variations inCCDN1,MYC,METandERBB2; and alterations in DNA methylation[2,5]. In addition, many more genes contribute to HCC progression, including Ras and other members of this superfamily of GTPases.
Proteins from Ras and Rho families belong to the Ras superfamily of GTPases and are closely related structurally. Based on their sequence homology and functionality, they are sorted into 5 families: Ras, Rho,Rab, Ran and Arf[6]. They act as molecular switches, cycling between inactive (GDP-bound state) and active(GTP-bound state) conformations. Guanine nucleotide Exchange Factors (GEFs) catalyze the release of GDP, favoring GTP binding. After the activation of effector pathways, GTP is rapidly hydrolyzed to GDP in a reaction accelerated by GAPs (GTPase activating proteins)[7]. Rho GTPases are also regulated by GDPdissociation inhibitors[6,8]. Although Ras proteins are more frequently mutated in human cancer[9],mutations in Rho family of GTPases, mainly Rho and Rac, have also been described[10-12]. In fact, Rho GTPase deregulation may contribute not only to cancer cell proliferation but also to invasion and metastasis[13].
In mammals, the Ras family comprises 36 members that regulate cell growth, differentiation and survival.Ras proteins are classified into 4 main subfamilies, namely Ras, Rap, Ral and R-Ras, each with several members[14]. They are activated by GEFs harboring a CDC25-homology domain that, together with the REM (Ras Exchange Motif) module, constitute the GEF-catalytic domain, which is conserved from yeast to humans[15]. Ras GEFs are classified into different protein families according to their specific, non-catalytic modules: C3G (RAPGEF1), PDZ-GEF1/2 (RAPGEF2/6), EPAC1/2 or cAMP-GEFI/II (RAPGEF3/4), MRGEF (RAPGEF5), SOS-1/2, Ras-GRF1/2, CalDAG-GEFII/I/III (RASGRP1/2/3), RasGRP4, RalGDS, RalGPS,
DOCK4 and PLC-epsilon-1 (PLCE1)[15-17].
Rho proteins (22 members in mammals) regulate actin cytoskeleton dynamics and vesicle trafficking[18].They are activated by GEFs from either Dbl family (with 69 members), which contain a DH (Dblhomology) catalytic domain and a PH (plekstrin homology) domain[15,19], or DOCK family (with 11 members), bearing a DHR2 (DOCK homology region 2) catalytic domain[20].
Both Ras GEF and Rho GEF families channel different signaling pathways that lead to the activation of specific GTPases, providing a fine-tuned regulation. In addition, their multi-domain structure allows complex autoregulatory mechanisms for the modulation of their target GTPases, some of which are still unknown. In the case of C3G, this mechanism has recently been uncovered[21]. Due to their function as specific GTPase activators, GEFs are susceptible nodes in the cell that modulate several signaling pathways such as MAPKs (ERKs, JNKs and p38 MAPKs) or PI3K. Consequently, their aberrant function is associated with a large number of human diseases, including cancer, where they may act as either promoters or suppressors of tumor growth and progression[22].
Some Ras and Rho proteins and their GEFs play specific roles in the liver. The cAMP/EPAC/Rap1 cascade is an important pro-survival pathway for hepatocytes with an inhibitory role in gluconeogenesis. In addition, EPAC1 acting through Rap1 protects from fibrosis by suppressing the activation and proliferation of hepatic stellate cells, while EPAC2/Rap1 plays a profibrotic role[23]. The EPAC2/Rap1 pathway also regulates lipid metabolism in the liver[24]. Moreover, Rap promotes proliferation during liver regeneration[23], and the Rho GEF, Ect2, also plays a role in liver regeneration[25].
The Rac GEF, Vav1, through a non-canonical p38MAPK pathway, decreases hepatocyte cell death during acute liver inflammation[26]. Another Rac GEF, P-Rex2, controls glucose homeostasis in the liver and inhibits PTEN in a Rac GEF-independent manner[27].
GEFs are deregulated in cancer through somatic mutations, changes in gene expression or post-translational modifications. Examples of Ras GEFs implicated in human cancer are RasGRF2, CalDAG-GEFI, CalDAGGEFII, RasGRP4 and Sos-1 (Ras subfamily); C3G, DOCK4 and PLC-ε-1(Rap subfamily); and RalGDS,Rgl1/2 and Rgr (Ral subfamily). Among Rho GEFs, aberrant activity of Mcf-2, Vav1-3, Bcr, ECT2,ARHGEF2/4/5/7/11/12/17, Trio, OBSCN, SPATA13, P-Rex1, P-Rex2a, Net1, AKAP13, Lfc, Tiam1, Ost, Clg(Dbl family) and DOCK1, 2/3/8/10 (DOCK family) has also been found in tumors[22,28-30].
Particularly, the deregulation of some of Ras and Rho GEFs has been associated with HCC progression and metastasis, justifying the growing interest. Here, we summarize the most remarkable data on this subject.
C3G AND OTHER RAS GEFS IN LIVER CANCER
C3G (Crk SH3-domain-binding guanine-nucleotide-releasing factor) is a GEF for Rap1 and other Ras proteins, such as R-Ras[31,32]. However, several C3G functions are not dependent on its GEF activity, but rather rely on its interaction with other proteins through its proline-rich domain and/or its ability to translocate to the nucleus[33-36]. There are two main C3G isoforms, A (the most common) and B, with 21 amino acids replacing 3 from the N-terminal domain[37]. C3G is essential for embryonic development[38]and regulates several cellular functions such as adhesion, migration, apoptosis and differentiation[37,39][Figure 1]. Its role in cancer depends on cellular context, tumor type and stage. C3G prevents malignant transformation induced by oncogenes in mouse fibroblasts[33,40]and its expression decreases in cervical squamous cell carcinoma[41]. In colon carcinoma (CRC), C3G plays a dual role, inhibiting migration and invasion, while favoring tumor growth[42]. C3G also reduces migration in highly invasive breast cancer cells[43]. In non-small-cell lung cancer, C3G levels increased[44], suggesting a role as a tumor promoter. In this line, p87C3G isoform (lacking the most N-terminal region) is upregulated in chronic myeloid leukemia(CML) and associated with this disease[45].
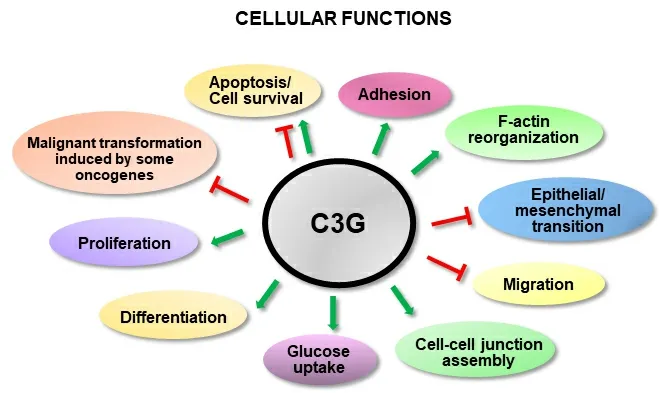
Figure 1. C3G cellular functions. The scheme shows the positive (green) or negative (red) regulation of different cellular functions by C3G.
Until very recently, there was no information in the literature supporting a role for C3G in liver cancer. The first data were obtained from different human cancer databases and indicate thatRAPGEF1mRNA levels increase in samples from HCC patients and in patient-derived xenografts, as compared to non-pathological liver samples[23]. Later, new analyses of cancer databases revealed thatRAPGEF1mRNA levels gradually increase in HCC patients as the disease progresses (from stages I to III), being also higher in HCC cell lines as compared to adult hepatocytes[46][Table 1]. This increase in C3G levels is associated with a lower survival[46], as is the presence of somatic mutations and other genetic alterations inRAPGEF1gene(amplification, deletion, etc.)[23]. C3G protein levels also increase in HCC cells, as compared to adult hepatocytes, and its downregulation by gene silencing reduces their tumorigenic properties, bothin vitroandin vivo[46]. However, the reduction in C3G levels enhances the pro-migratory and pro-invasive capacity of HCC cells by favoring the acquisition of a more mesenchymal phenotype. Hence, low levels of C3G correlate with lung metastasis, although its growth is associated to the recovery of a highC3Gexpression.Moreover, C3G is required for the correct activation of HGF/MET signaling in HCC cells[46]. All these data indicate that C3G plays a key role in HCC, promoting tumor growth, and the regulation of its levels may facilitate HCC growth and progression. However, the potential contribution of the main C3G target, Rap, to these actions of C3G remains unclear.
Little is known about Rap function in HCC and the available data are controversial. Transfection of Rap1 decreases proliferation of Hep3B cells by reducing ERKs activation and suppresses tumor growth[47]. In contrast, Rap1B is upregulated in HBV-induced HCC, promoting proliferation and migration[48]. More recent studies also revealed an upregulation of eitherRAP2B[49]orRAP1Bexpression[50]in human HCC samples and in some cell lines, which leads to an increased proliferation, tumor growth and migration. In addition, our analyses of data from HCC patient samples obtained from TCGA database also show no changes inRAP2C, but an increase inRAP1A/1B/2A/2BmRNA levels that reaches statistical significance in the case ofRAP2A, as compared to non-pathological samples [Figure 2]. This is associated with a significant reduction in the overall survival, while no differences in disease-free survival are observed.RAP1A/1B/2A/2B/2Cgenes also present amplifications or other genetic alterations in HCC [Figure 3].
Table 1. Ras and Rho GEFs involved in primary liver cancer
The effects of C3G and Rap proteins are not equivalent, suggesting that C3G might have Rap-dependent and -independent actions in HCC. Other Rap GEFs such as EPAC1/2 could contribute to regulate Rap activity. Although there are no data about them, EPAC1/2 regulate liver fibrosis[51][Table 1], a previous step towards HCC development.
Concerning the potential role of other Ras GEFs in HCC,RASGRP1is upregulated in HCC patient samples and human HCC cell lines, promoting proliferation[52].RASGRP3andPLCE1are also upregulated[53]. Other Ras GEFs, such asRALGPS2orRGL1, could play a role, based on genetic alterations found in TCGA[Figure 3].
In CCA, the function of C3G and/or Rap remains unknown. Using data from a TCGA cohort of patients with CCA, we found a significant increase in the mRNA levels of C3G (RAPGEF1),RAP1A/1BandRAP2A/2B, but not ofRAP2C, in tumors as compared to non-pathological liver samples [Figure 4] and genetic alterations [Figure 3]. Surprisingly, in contrast to HCC data, the increased levels of RapGEF1 and Rap family members are not associated with a lower overall survival. These results reinforce the idea that,although hepatocytes and cholangiocytes share a common liver bipotential progenitor, HCC and CCA are clinically two distinct entities in terms of mechanisms, diagnosis and treatment[54]. Hence, more studies are necessary to uncover the specific characteristics of CCAs. Additionally, the limited number of CCA samples available in the TCGA-CHOL dataset can bias result interpretation.
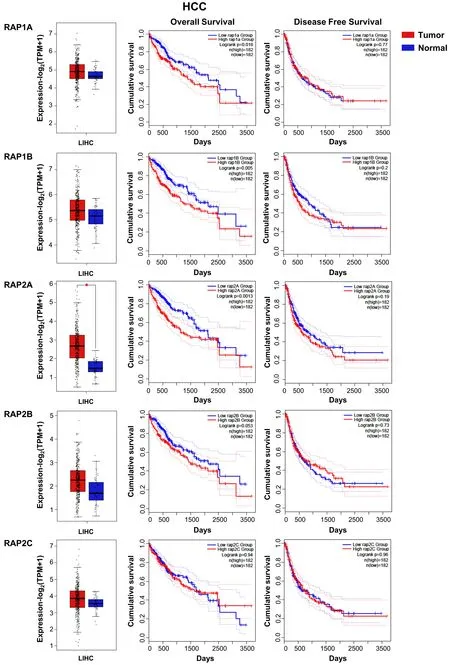
Figure 2. RAP GTPases mRNA expression in HCC patients and its relationship with survival. From top to bottom, RAP1A, -1B, -2A, -2B and -2C: Box plots (left) show mRNA levels expressed as log2 (TPM + 1) in tumor (red) and adjacent liver non-pathological (blue)samples. Kaplan-Meier curves show overall survival (middle) and disease-free cumulative survival (right), comparing patients with high(red) vs. low (blue) expression levels for each analyzed gene (median TPM cutoff was chosen). All data were generated through GEPIA2 portal (Gene Expression Profiling Interactive Analysis; http://gepia2.cancer-pku.cn/#index). Log rank value is indicated for each curve. GEPIA2 mRNASeq dataset analyses are based on UCSC Xena project (http://xena.ucsc.edu) TCGA-LIHC [Liver Hepatocellular Carcinoma (HCC)] cohort for (50 normal and 371 tumor samples) patients. Results are considered statistically significant for P values ≤ 0.05 (*).
Regarding the potential regulation of C3G expression by miRNAs, it is important to mention that, although bioinformatic tools, such as TargetScanHuman database (www.targetscan.org), predict potential interactions of different miRNAs withRAPGEF1gene, there are no publications validatingin vitroorin vivothe existence of these interactions in any model. However, miR-27a, which reduces viral replication and infectivity of HBV in human hepatoma cells, can repressRAPGEF2in Huh7.5 HCC cells[55].
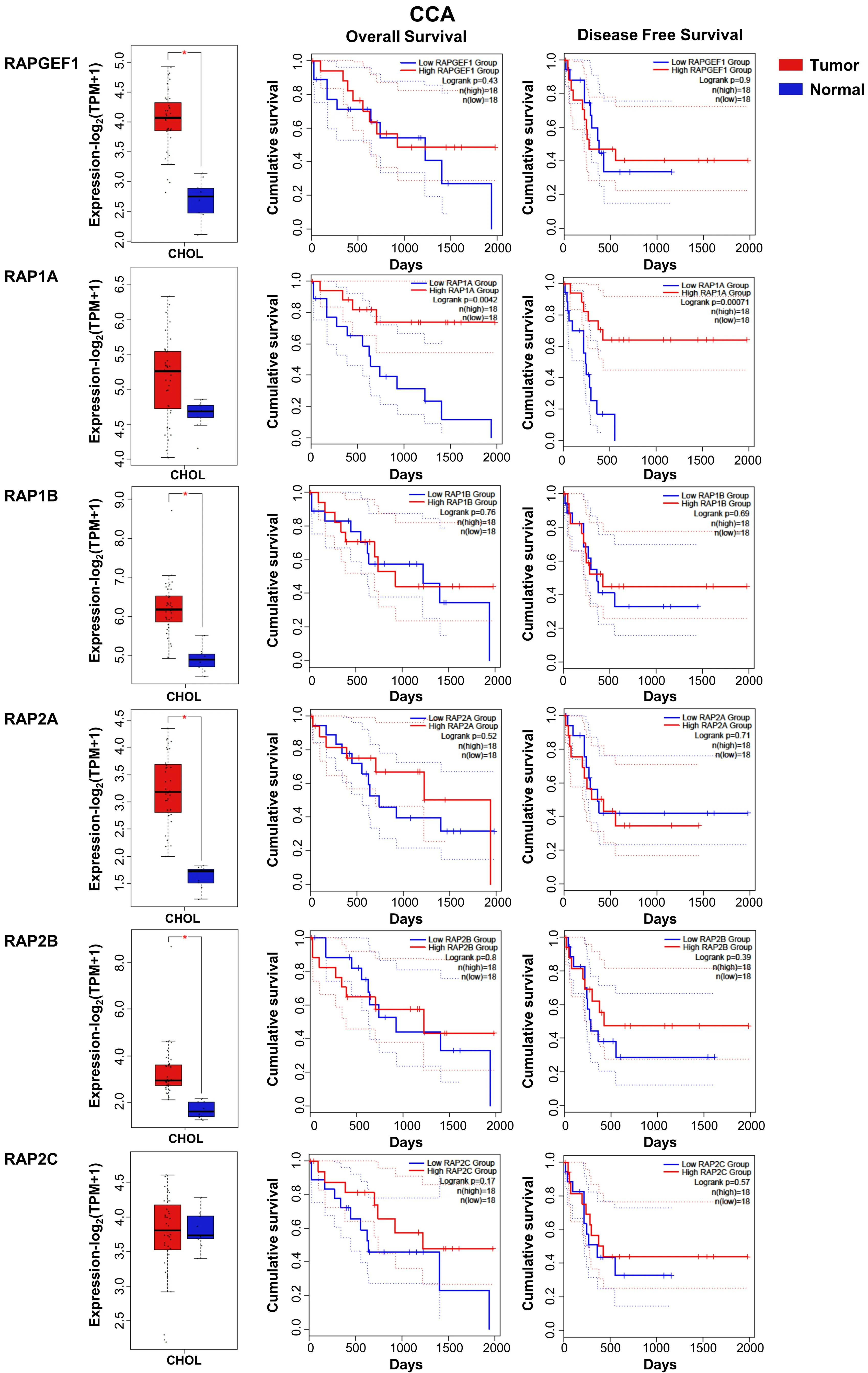
Figure 4. RAPGEF1 and RAP GTPases mRNA expression in CCA patients and its relationship with survival. From top to bottom, RAPGEF1,RAP1A, -1B, -2A, -2B and -2C: Box plots (left) show mRNA levels expressed as log2 (TPM + 1) in tumor (red) and adjacent liver nonpathological samples (blue) samples. Kaplan-Meier curves show overall survival (middle) and disease-free cumulative survival (right),comparing patients with high (red) vs. low (blue) expression levels of each analyzed gene (median TPM cutoff was chosen). All data were generated through GEPIA2 portal (Gene Expression Profiling Interactive Analysis; http://gepia2.cancer-pku.cn/#index). Log rank value is indicated for each curve. GEPIA2 mRNASeq dataset analyses are based on UCSC Xena project (http://xena.ucsc.edu) TCGACHOL (9 normal and 36 tumor samples) from patients. Results are considered statistically significant for P values ≤ 0.05(*). CCA:Cholangiocarcinoma.
CRK, A C3G BINDING PROTEIN WITH A ROLE IN HCC
CRK (CT10 regulation of Kinase) proteins are adaptors involved in the activation of several signaling pathways[57]. This family of proteins encompasses two alternative spliced isoforms, CRKI and CRKII, and the related CRKL [v-crk sarcoma virus CT10 oncogene homolog (avian)-like] protein. They are ubiquitously expressed and conserved throughout eukaryotes. CRK proteins harbor one N-terminal Src homology 2 (SH2) domain and one (in CRKI) or two (in CRKII and CRKL) C-terminal Src homology 3(SH3) domain[58], which are essential to interact with a great number of signaling molecules such as Sos,Gab1, Bcr-Abl, p130Cas and C3G. This allows the integration of a great variety of signals, leading to the regulation of several cellular functions. In cancer, CRK proteins play important roles. CRKL is overexpressed in a number of cancers such as gastric cancer, glioblastoma, lung cancer, CML and HCC[59].Moreover, in breast cancer patients, high soluble CRKL levels are found in the serum, associated with advanced stages of the disease[60].
CRKL has been proposed as a prognostic biomarker for HCC based on its high expression in patient samples and its inverse correlation with the overall survival of HCC patients[61]. In addition, CRKL promotes migration in HCC cells. Several studies have demonstrated that CRKL is overexpressed in HCC, inducing migration, invasion and proliferation[62-64]. In agreement with this, our analysis using TCGA data revealed an increase inCRKLmRNA levels in HCC patient samples as compared to control liver [Figure 5]. Moreover,highCRKLlevels are associated to a lower survival. InCCA,CRKandCRKLexpression is increased, being statistically significant forCRKL[Figure 5].
Some of the mechanisms responsible for CRKL regulation have been uncovered. It is noticeable thatCRKLexpression is tightly regulated by different miRNAs in the liver and this regulation is altered in HCC.Hence, p53-induced miRNA-215 downregulates CRKL, acting through the long non-coding RNAPCAT-1,which decreases proliferation, tumor growth and migration/invasion of hepatocytes[63]. However, in HCC cells, this mechanism fails. Similarly, miR-429 negatively regulatesCRKLexpression post-transcriptionally,a mechanism altered in HCC, leading to an increased migration and invasion mediated by CRKL upregulation[62]. ERKs activation and epithelial-mesenchymal transition (EMT) contribute to these actions of the miR-429-CRKL axis. Furthermore, the upregulation in HCC of the transcription factor E-Twenty-Six variant gene 6, a negative regulator ofmiR-429expression, leads to CRKL overexpression and enhances migration and invasion[64]. Additionally, CRKL downregulatesmiR-429expression. On the other hand, the long non-coding RNA AFAP1-AS1 (Actin filament-associated protein 1 antisense RNA 1), upregulated in HCC, also induces HCC proliferation, migration and invasion through increasing CRKL levels, leading to a higherAFAP1-AS1expression[65]. In contrast to all this, a previous work showed that CRKL overexpression decreasedin vitroproliferation, migration and invasion in the murine hepatocarcinoma Hca-P cell line[66].
Another member of the CRK family, CRKII, could also contribute to promote HCC progression based on the increased proliferation, migration and invasion induced by CRKII overexpression in Hca-P cells[67]. In agreement with this, the induced upregulation of CRKI/II and Rac1 by Annexin 5 in HCC mediates proliferation and invasion of Hca-P cells and xenograft tumor growth[68].
In CCA, data from TCGA indicate the existence of deep deletions in CRK and missense mutations in CRKL[Figure 3].
In conclusion, although CRK is a C3G partner, its role in HCC is not necessarily associated with C3G, but it may depend on complex interactions with different proteins.
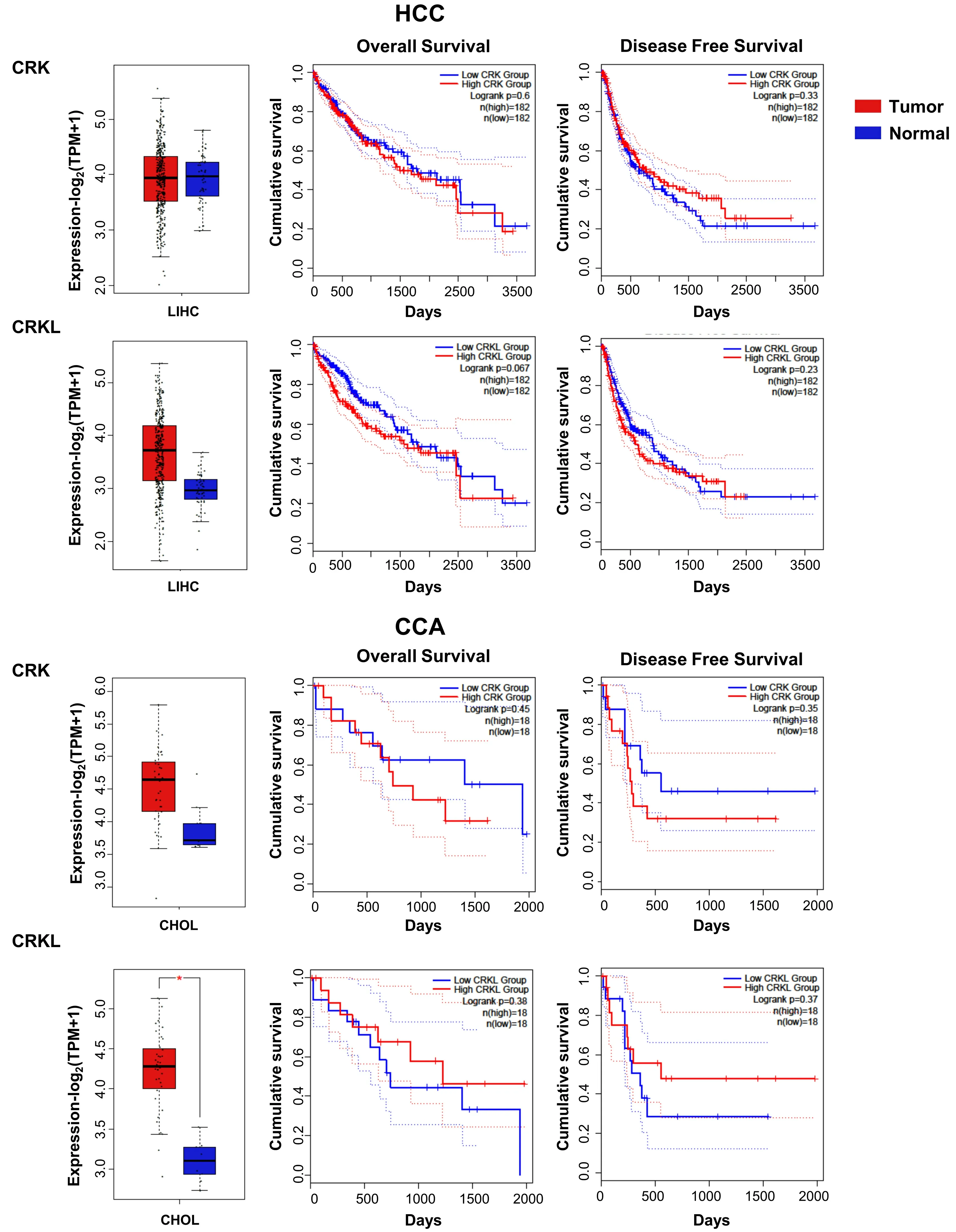
Figure 5. CRK and CRKL mRNA expression in HCC and CCA patients and its relationship with survival. Box plots (left) show CRK and CRKL mRNA levels expressed as log2 (TPM + 1) in tumor (red) and adjacent liver non-pathological samples (blue) samples. Kaplan-Meier curves show overall survival (middle) and disease-free survival probability (right) comparing patients with high (red) vs. low(blue) expression levels of each analyzed gene (median TPM cutoff was chosen). All data were generated through GEPIA2 portal (Gene Expression Profiling Interactive Analysis; http://gepia2.cancer-pku.cn/#index). Log rank value is indicated for each curve. GEPIA2 mRNASeq dataset analyses are based on UCSC Xena project (http://xena.ucsc.edu) TCGA-LIHC and TCGA-CHOL, for HCC (50 normal and 371 tumor samples) and CCA (9 normal and 36 tumor samples) patients, respectively. Results are considered statistically significant for P values ≤ 0.05(*). HCC: Hepatocellular carcinoma; CCA: cholangiocarcinoma.
RHO GEFS IN LIVER CANCER
As mentioned above, alterations of Rho and Ras GEFs have been frequently reported in liver cancer, being protein levels upregulation or hyperactivity the most reported events. In addition, we found several genetic alterations in our analyses of databases in both HCC and CCA [Figure 6]. We summarize here the most significant Rho GEFs involved in liver cancer [Table 1].
Tsar Vyslav rose up in his place and questioned Helen the Beautiful and she related to him the whole: how Tsarevitch Ivan had won her, with the Horse with the Golden Mane and the Fire Bird, and how his two elder brothers had slain him as he lay asleep and had threatened her with death so that she should say what they bade.
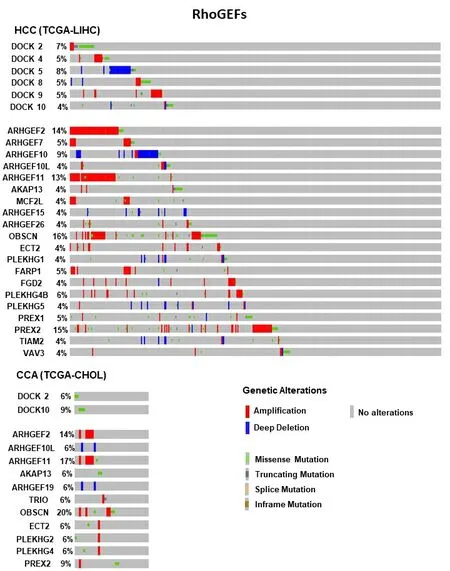
Figure 6. Frequent genetic alterations in Rho GEFs genes in HCC and CCA. The percentage of patients (when ≥ 4%) with genetic alterations for genes from Rho GEF family are shown. The type of these alterations is indicated by the color, as specified in the legend.Mutation data from whole exome sequencing were taken from cBioportal (https://www.cbioportal.org/) from Liver Hepatocellular Carcinoma (TCGA, Firehose Legacy, 442 samples) and Cholangiocarcinoma (TCGA, Firehose Legacy, 51 samples). HCC:Hepatocellular carcinoma; CCA: cholangiocarcinoma.
Tiam1 (T-cell lymphoma invasion and metastasis 1) protein is a Rac GEF that belongs to Dbl protein family[69]. It was identified as an invasion- and metastasis-promoting gene in a murine T-lymphoma cell line[70]. Tiam1 activates Rac1, inducing migration, invasion and metastasis of many tumor cells[71,72].Furthermore, Tiam1 has a significant role in promoting tumor progression in a variety of cancers, such as breast cancer, CRC and lung cancer[13,73,74]. Tiam1 is much more strongly expressed in almost all HCCs than in normal liver and cirrhotic liver tissues[75]. Furthermore,Tiam1andRac1expression is upregulated in HCC, which correlated with advanced clinical stages[75-77]and a significant reduction in disease-specific survival[75]. However, the precise molecular mechanisms of Tiam1 actions in HCC tumorigenesis are still unknown. Overexpression of Tiam1 increases proliferation, migration and invasion in HCC cell lines[77].
Moreover,in vivofunctional studies showed that Tiam1 upregulation enhances tumorigenicity and metastatic potential, while Tiam1 knockdown delays tumor growth and inhibits metastases formation[77]. A recent report also showed thatTiam1expression can be epigenetically regulated in HCC by KDM6B demethylase leading to invasion and metastasis[78,79]. MicroRNAs also regulate HCC migration and proliferation by modulatingTiam1expression[80,81]. Interestingly,Tiam1expression is also increased in CCA, correlating with a higher degree of malignancy[82]. Furthermore, Tiam1 inhibition prevented CCA cell proliferation and migration[82].
TIAM2gene is a homolog ofTIAM1, a Rac GEF with important roles in neuron development and human malignancies[69]. Tiam2 short form (TIAM2S) is upregulated at both protein and mRNA levels in HCC[83].Posttranscriptional regulation, mainly achieved by SP1 transcription factor, inducesTIAM2SmRNA expression in HCC cell lines[84]. TIAM2S promotes cell growth and invasion through EMT regulation, being associated with a metastatic phenotype of HCCs[83].
ARHGEF10L (Rho guanine nucleotide exchange factor 10-Like) is a specific GEF for RhoA. ARHGEF10L is upregulated in HCC, especially at stage III[85], promoting HCC proliferation and invasion through activation of the RhoA-ROCK1-ERM (ezrin/radixin/moeisin) pathway and EMT induction[85].
ARHGEF39 (or C9orf100) is a member of the human Dbl family of Rho GEFs[86]. ARHGEF39 mRNA and protein levels are upregulated in HCC samples compared to non-pathological adjacent liver tissue[87,88], and its expression correlates with worse prognosis[87]. ARHGEF39 promotes cell proliferation and migration in HCC cell lines[88].
Another ARHGEF, ARHGEF19, promotes HCC cell proliferation and invasion[89]. Interestingly, the microRNA miR-503 inhibits HCC invasion and metastasis through inhibition ofARHGEF19expression[89].
ARHGEF9 is a Rho GEF able to activate the Rho GTPase Cdc42. CHD1L (Chromodomain helicase/ATPase DNA binding protein 1-like gene) induces ARHGEF9 transcription, leading to Cdc42 activation, which promotes filopodia formation and EMT induction, enhancing HCC invasion and metastasis generation[90].Hence, overexpression of ARHGEF9 positively correlates with CHD1L upregulation and poor disease-free survival[90].
ECT2 (Epithelial cell transforming sequence two protein) is significantly associated with early recurrent HCC disease and poor survival[91,92]. Its knockdown prevents the activation of Rho/ERKs signaling, enhances apoptosis and reduces migration and invasion of HCC cells[91]. Furthermore, the microRNA miR-490-5p reduces HCC metastasis and stemness through ECT2 inhibition[92,93]. In addition, in CCA cells, miR‐194 promotes apoptosis and inhibits proliferation and migration through ECT2 downregulation and Rho signaling blockade[94].
Lbc (Lymphoid blast crisis, or AKAP13) is a Rho GEF[69], originally isolated as an oncogene with increased GEF activity[95]. Lbc is not expressed in adult liver under physiological conditions. However, it is upregulated in HCC[96]. Lbc increases HCC cell growth and inducesBcl-2expression and BAD phosphorylation, which is involved in the generation of HCC resistance to doxorubicin[96].
VAV2/3 are GEFs for RhoA, RhoG and Rac1 that modulate their activity, which is important for HCC cell migration.VAV3expression its downregulated in HCC by the transcription factor KLF6. Thus, KLF6 depletion increases Rac1 activity in a VAV3-dependent manner[97]. VAV2 promotes Rac1 and Cdc42 activation, leading to lamellipodia formation, facilitating metastasis of HCC cells. Interestingly, miRNA‐195 suppresses angiogenesis and metastasis of HCC by inhibiting the expression ofVEGF,VAV2andCdc42[98].
Several other GEFs are upregulated in HCC and might play a role as potential diagnosis and prognosis biomarkers of this cancer. Among them, FGD1, GEF-H1 and NET1 from Rho-GEF family can be highlighted[53,99-104].
In contrast to Rho GEFs, Rho GAPs, such as DLC1 (Deleted in Liver Cancer 1) and DLC2, act as tumor suppressors in HCC.DLC1gene is inactivated in hepatocarcinogenesis[105], suppressing cell proliferation and migration[106]. DLC2 expression is also downregulated in HCC[107,108], which is associated with cell differentiation and poor prognosis[108].
CONCLUSION
GEFs are frequently deregulated and appear to be essential mediators in liver cancer. They may represent diagnosis biomarkers and attractive targets for liver cancer therapy. However, future studies are needed to establish their precise role in liver cancer. This would allow determining the potential relevance of targeting GEFs in HCC treatment, probably through the design of inhibitors for specific domains, as some of these proteins can act through GEF dependent and independent mechanisms.
DECLARATIONS
Authors’ contributions
All the authors contribute to the writing of this review.
Performed the analysis of patient data from TCGA database: Sequera C Coordinated all the work: Porras A
Availability of data and materials
Not applicable.
Financial support and sponsorship
The work was supported by grants from the Spanish Ministry of Economy and Competitiveness (SAF2016-76588-C2-1-R and PID2019-104143RB-C22 to Porras A; and SAF2016-76588-C2-2-R and PID2019-104143RB-C21 to Guerrero G and PID2019-104991RB-I00 to Bragado P), and by two grants from the Council of Education of Junta de Castilla y León, Spain (SA017U16 and SA078P20 to Guerrero C). All funding was cosponsored by the European FEDER Program. Sequera C was supported by a fellowship from Complutense University from Madrid. Gutierrez-Uzquiza A is supported by Madrid Community Program for Talent Attraction (MRF 2017-T1/BMD-5468). Bragado P received support from BBVA (Becas Leonardo 2018, BBM-TRA-0041).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
? The Author(s) 2021.

- Hepatoma Research的其它文章
- GENERAL INFORMATION
- AUTHOR INSTRUCTIONS
- Surgical perspective on treatment of pediatric undifferentiated sarcoma of the liver
- The prognostic evaluation of marginal positive resection in hepatoblastoma: Japanese experience
- Role of CD4+ T-cells in the pathology of non-alcoholic fatty liver disease and related diseases
- Defining the role of laparoscopic liver resection in elderly HCC patients: a propensity score matched analysis