
HybridSucc: A Hybrid-learning Architecture for General and Species-specific Succinylation Site Prediction
2020-11-06 01:04:10WanshanNingHaodongXuPeiranJiangHanChengWankunDengYapingGuoYuXue
Wanshan Ning, Haodong Xu, Peiran Jiang, Han Cheng, Wankun Deng,Yaping Guo, Yu Xue,3,*
1 Department of Bioinformatics and Systems Biology, Key Laboratory of Molecular Biophysics of the Ministry of Education, Hubei Bioinformatics and Molecular Imaging Key Laboratory, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan 430074, China
2 School of Life Sciences, Zhengzhou University, Zhengzhou 450001, China
3 Huazhong University of Science and Technology Ezhou Industrial Technology Research Institute, Ezhou 436044, China
KEYWORDS Lysine succinylation;Post-translational modification;Deep-learning;Machine-learning;Deep neural network
Abstract As an important protein acylation modification,lysine succinylation (Ksucc) is involved in diverse biological processes, and participates in human tumorigenesis. Here, we collected 26,243 non-redundant known Ksucc sites from 13 species as the benchmark data set,combined 10 types of informative features, and implemented a hybrid-learning architecture by integratingdeep-learning and conventionalmachine-learningalgorithms into a single framework. We constructed a new tool named HybridSucc, which achieved area under curve(AUC) values of 0.885 and 0.952 for general and human-specific prediction of Ksucc sites,respectively. In comparison,the accuracy of Hybrid-Succ was 17.84%–50.62%better than that of other existing tools.Using HybridSucc,we conducted a proteome-wide prediction and prioritized 370 cancer mutations that change Ksucc states of 218 important proteins, including PKM2, SHMT2, and IDH2. We not only developed a high-profile tool for predicting Ksucc sites, but also generated useful candidates for further experimental consideration.The online service of HybridSucc can be freely accessed for academic research at http://hybridsucc.biocuckoo.org/.
Introduction
In proteins,positively charged lysine(Lys)residues are preferentially subject to a broad spectrum of post-translational modifications (PTMs), especially a number of short-chain Lys acylations such as Lys acetylation(Kac)and Lys succinylation(Ksucc) [1–4]. Biochemically, Kac attaches a small and hydrophobic acetyl group to the amine group of the Lys residue to neutralize its positive charge,with a mass of 42.0106 Da[1,2]. Ksucc adds a bulkier and acidic succinyl group to alter the Lys charge from +1 to -1, with a much larger mass of 100.0186 Da[1,2,5].Generally,Ksucc is comparable to protein phosphorylation, a well-studied PTM that also induces a -1 charge on proteins by adding a 79.9663-Da phosphate group to a serine, threonine, or tyrosine residue [2,3]. Since protein phosphorylation is involved in almost all biological processes by dramatically changing the structure, enzymatic activity,and stability of the proteins, it has been proposed that Ksucc might also be functionally important [2,3]. Similar to Kac,Ksucc occurs in both histone and non-histone proteins, and participates in regulating metabolism [6,7], immunity [8],autophagy [9], genome stability [10], and gene expression[11]. The dysregulation of Ksucc is highly associated with human diseases such as cancer and neurodegenerative disorders [2,12]. Thus, identification of modified substrates with exact Ksucc sites is fundamental for understanding the molecular mechanisms and regulatory roles of Ksucc.
In 1961, Ksucc was initially developed as a biochemical assay to efficiently test wheal-and-erythema responses via inhibition of antibody formation in serum [13]. 50 years later,Ksucc was discovered to naturally occur on protein Lys residuesin vivo,as a novel PTM[5,14].Rapid progresses in development of the state-of-the-art techniques for succinylomic profiling have led to the detection of hundreds or thousands of Ksucc sites through high-throughput mass spectrometry and immunoaffinity enrichment of Ksucc peptides using antisuccinyllysine antibody. In 2011, Dr. Yingming Zhao’s group conducted a pilot analysis of the Lys succinylome, and identified 69 Ksucc sites of 14 proteins inEscherichia coli[5]. Later,they carried out a more comprehensive profiling and detected 2565 Ksucc sites in 779 proteins from mouse liver and cell lines[7]. As more and more experimental studies were performed,the collection, integration and annotation of known Ksucc substrates and sites emerged as important topics for sharing and reusing data. In 2014, we developed a database named compendium of protein lysine modifications (CPLM) 2.0, by manually collecting known substrates and sites for 12 types of protein Lys modifications(PLMs),including 897 Ksucc substrates with 2523 sites [4]. Later, CPLM 2.0 was updated into protein lysine modification database (PLMD) 3.0 for 20 types of PLMs,containing 18,593 non-redundant Ksucc sites in 6377 proteins [15]. In addition, the most popular protein phosphorylation data resource, PhosphoSitePlus, also curated 4627 Ksucc sites [16].
Publicly available databases contain high-quality data sets for training computational models with various algorithms,which provided an alternative means for identification of potential Ksucc sites from protein sequences. In April 2015,Zhao et al. reported the first tool, SucPred, for the prediction of Ksucc sites[17].They took known Ksucc sites from CPLM 2.0 [4], adopted multiple sequence features including autocorrelation function(ACF),2 types of physicochemical properties of amino acids, and pseudo amino acid composition(PseAAC), and used the support vector machine (SVM) algorithm for training [17]. After 4 months, we released an SVMbased tool named SuccFind, in which PseAAC, composition ofk-spaced amino acid pairs (CKSAAP), and 544 types of physicochemical properties maintained in the Amino Acid index database (AAindex) were combined as predictive features [18]. In 2018, Chen et al. used orthogonal binary coding(OBC) and AAindex encodings to develop a novel deeplearning framework,MUscADEL,for an improved prediction of 8 types of PLMs including Ksucc [19]. To date, 13 site predictors have been designed,and multiple types of sequence and structural features have been used (Table S1). However, it is unclear which features are the most informative for predicting Ksucc sites. Also, most of the tools were implemented in conventional machine-learning algorithms, which are less efficient in feature representation than deep-learning algorithms. The extent to which deep-learning algorithms can improve the prediction accuracy remains to be probed.In addition to the general prediction of Ksucc sites, only SuccinSite2.0 constructed seven species-specific predictors [20]. Due to the data accumulation, more organisms should be included for the prediction of species-specific Ksucc sites.
In this work, we compiled a non-redundant data set, containing 26,243 experimentally identified Ksucc sites of 8830 proteins from 13 organisms, includingHomo sapiens,Mus musculus,Rattus norvegicus,Saccharomyces cerevisiae,Oryza sativa,Brachypodium distachyon,Solanum lycopersicum,Toxoplasma gondii,E. coli,Vibrio parahaemolyticus,Bacillus subtilis,Corynebacterium glutamicum, andMycobacterium tuberculosis(Figure 1 and Table S2). After homology clearance, we carefully evaluated 7 types of sequence-derived features including the PseAAC, CKSAAP, OBC, AAindex,ACF, Group-based Prediction System (GPS), and positionspecific scoring matrix(PSSM),as well as 3 types of structural features including the accessible surface area(ASA),secondary structure, (SS) and backbone torsion angles (BTA) (Figure 1 and Table S3) [17,18,20–23]. Three conventional machinelearning algorithms, including penalized logistic regression(PLR), SVM, and random forest (RF), were adopted to train computational models on the benchmark data set separately for each feature. The accuracy of the general or speciesspecific prediction of Ksucc sites was evaluated, whereas the results demonstrate that the 10 types of features were all informative (Table S4). We also implemented a deep neural network (DNN) framework and compared the predictions to the PLR algorithm. Surprisingly, we found that deeplearning and conventional machine-learning algorithms exhibited strikingly different advantages for representing distinct features (Table S4). Then, we merged DNN and PLR into a hybrid-learning architecture, and developed the HybridSucc predictor (Figure 1). In comparison, HybridSucc significantly outperformed other existing tools, and achieved a ≥17.84%improvement of the area under curve (AUC) value (0.885vs.0.751)for the general prediction of Ksucc sites.Using Hybrid-Succ, we conducted a proteome-wide prediction, and prioritized 5251 known and 3615 predicted Ksucc sites to be potentially functional (Figure 1 and Table S5). Moreover, we also mapped cancer mutations in The Cancer Genome Atlas(TCGA) [24] to human Ksucc substrates, defined Ksuccrelated mutations (KsuMs), and developed a new statistical approach of the gradual distribution of probability density(GDPD)to estimate the impact of cancer mutations on Ksucc sites. We identified 370 highly potential KsuMs in 218 genes,including a number of well-studied genes involved in tumorigenesis such as the genes encoding pyruvate kinase M2 (PKM2)[25], serine hydroxymethyltransferase 2 (SHMT2) [12], and isocitrate dehydrogenase 2 (IDH2) [26] (Figure 1 andTable S6). Taken together, our study not only established a novel hybrid-learning architecture to achieve a superior accuracy for the prediction of Ksucc sites, but also systematically characterized human KsuMs that potentially function in cancer.
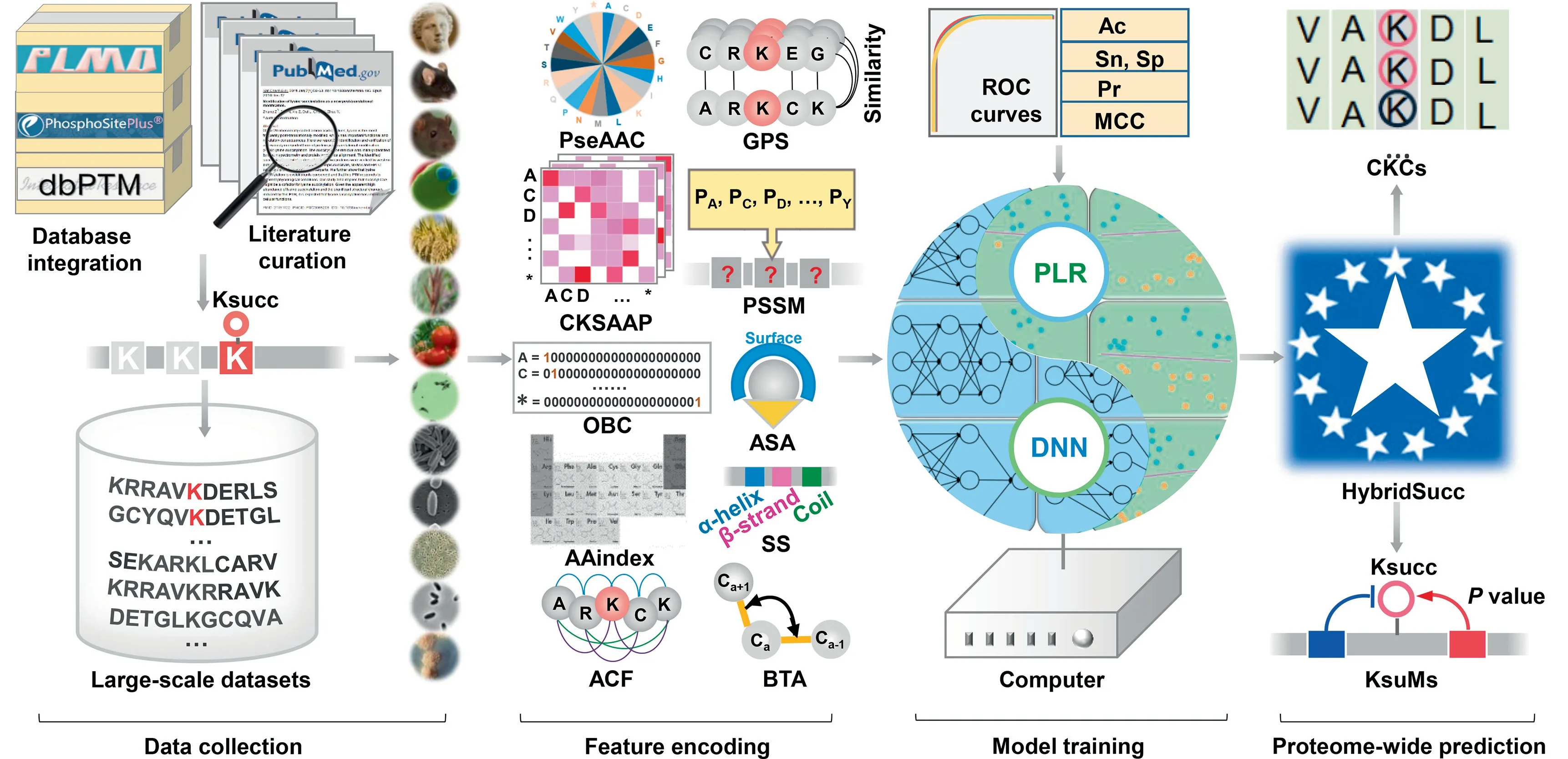
Figure 1 Experimental procedure of the study
Method
Data collection and preparation
From 3 public databases, PLMD 3.0 [15], PhosphoSitePlus[16], and dbPTM [27], we obtained 18,593 non-redundant known Ksucc sites.To avoid missing any data,we further conducted a literature curation by searching PubMed with multiple keywords such as ‘‘protein succinylation”, ‘‘lysine succinylation” and ‘‘succinylated protein”, and manually collected 21,392 experimentally identified Ksucc sites. The two data sets were merged, and we mapped the Ksucc sites to primary protein sequences downloaded from the UniProt database [28] to pinpoint the exact Ksucc positions for each species. We obtained 26,243 non-redundant Ksucc sites in 8830 proteins of 13 organisms, including eight eukaryotes,H. sapiens,M. musculus,R. norvegicus,O. sativa,S. lycopersicum,B. distachyon,T. gondiiandS. cerevisiae, and five prokaryotes,E. coli,V. parahaemolyticus,B. subtilis,C. glutamicum, andM. tuberculosis.
Prior to preparation of the benchmark data sets for training and testing, homologous sites were cleared using the CD-HIT program to avoid overfitting [29], with a threshold of 40%sequence similarity [22]. If two Ksucc proteins are modified at the same positions with a >40% sequence identity, only one of them was reserved. Then, we defined a Ksucc site peptide KSP (m,n) as a Lys residue flanked bymresidues upstream andnresidues downstream. Because too many parameters required fine-tuning in this study, we adopted KSP(10,10)to enable rapid training.As previously described[22], KSP (10, 10) peptides around known Ksucc sites were regarded as positive data, whereas KSP (10, 10) items derived from the remaining non-succinylated Lys residues in the same proteins were taken as negative data. The redundancy at the peptide level was cleared for positive and negative data,respectively,and only one KSP(10,10)was reserved if multiple identical peptides were detected. For the general prediction of Ksucc sites,the non-redundant data set for training and testing contained 21,770 positive sites and 165,071 negative sites from 7415 substrates, respectively (Table S2). For the speciesspecific predictions, the same redundancy clearance procedure was performed to obtain the benchmark data set for each species(Table S2).All benchmark data sets of known Ksucc substrates with UniProt accession numbers, protein sequences,Ksucc positions, organisms, and PubMed IDs (PMIDs) of original references, can be downloaded at http://hybridsucc.biocuckoo.org/download.php.
Feature encoding scheme
For each benchmark data set, 10 types of features were separately extracted from the KSP (10, 10) peptides of both positive and negative data sets, including 7 types of sequencederived features, PseAAC, CKSAAP, OBC, AAindex, ACF,GPS, and PSSM, as well as 3 structural features, ASA, SS,and BTA [17,18,20–23] (Table S3). The 10 types of features were defined as: (1) PseAAC, which denotes the amino acid frequencies [17,18,20–23]; (2) CKSAAP, which indicates the composition of amino acid pairs separated bykother residues[20,21]; (3) OBC, which denotes position-specific amino acids[20]; (4) AAindex, a database that contains 566 amino acid indices of physicochemical properties [17,18,20–23]; (5) ACF,which represents the sequence order and correlation information[17];(6)GPS,which reflects the position-weighted similarity of amino acids [22]; (7) PSSM, which provides the probability of an amino acid occurrence at a specific position[20,23];(8)ASA,which indicates the exposed area of an amino acid residue to solvent[23];(9)SS,which represents 3 types of structural elements, including α-helix, β-strand, and coil [23];(10) BTA, which offers continuous angle information of the local conformation of proteins,including the backbone torsion angles φ and Ψ,the angle between Cαi-1-Cαi-Cαi+1(θ)and the dihedral angle rotated about the Cαi-Cαi+1bond (τ) [23].More details on the implementation of the 10 features were described in File S1.
Conventional machine-learning algorithms
In this study,3 classical machine-learning algorithms including PLR[30],SVM[21],and RF[20]were used to evaluate the predictive capacities of the 10 features for general or speciesspecific prediction of Ksucc sites. For the PLR, SVM and RF algorithms,five measurements of accuracy(Ac),sensitivity(Sn), specificity (Sp), precision (Pr) and Matthews correlation coefficient (MCC) were calculated as:

For each algorithm,4-,6-,8-,and 10-fold cross-validations were performed separately.The receiver operating characteristic (ROC) curves were illustrated for Snvs.1-Sp scores and the AUC values were calculated. For accurate estimation of the performance, the 10-fold cross-validation was independently performed 100 times and the average AUC was calculated for each algorithm.
To further improve accuracy and prevent overfitting, we refined the original PLR algorithm by adding two steps of random mutation and random zeroing. First, the weights of different features were calculated by PLR with the least absolute shrinkage and selection operator (LASSO) penalty[30], and the 10-fold cross-validation AUC was computed.Then we selected an initial weight of +1 or -1 per time,and recalculated the AUC. The manipulation was adopted if the AUC score increased, and the random mutation process was stopped when the AUC was not enhanced any longer.To avoid the local optimization, we added a step by random zeroing one weight,and re-conducted the multi-round random mutation process. Such a procedure was iteratively repeated until convergence was reached.
Deep-learning algorithm
A 4-layer DNN framework was implemented, and each layer consisted of a number of computational units called neurons(Table S7). To avoid over-fitting, Dropout was used by randomly dropping nodes from the two hidden layers if the accuracy increased.In each layer,all neurons constitute an internal feature representation, which also acts as the output of the layer. In the first step, the input layer receives data matrices,in which each line represents a unique KSP (10, 10) peptide,whereas columns contain numerical data generated by various feature encoding methods.For each neuron of the input layer,a data matrixxis transformed by a rectified linear unit(ReLU)activation function, which is defined as:

The first hidden layer is mainly adopted for feature extraction and representation, and the second is a fully connected hidden layer for generating predictions. TheReLUactivation function is used for each node. The output layer contains twosigmoidneurons to calculate aPsuccscore for a given KSP (10, 10) peptidey, defined as:
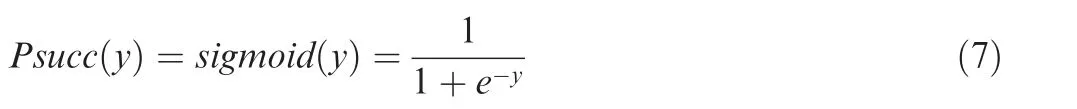
ThePsuccvalue,ranging from 0 to 1,denotes the probability of a KSP(10, 10)to be a real Ksucc site. Our implementation utilized the Keras 2.0.4 library (http://github.com/fchollet/keras) with the tensorflow 1.2.0 backend, which was configured in the graphics processing units (GPUs) of the NVIDIA CUDA development environment for parallel computing. During the training, transient parameters such as the learning rate, degree of momentum, mini-batch size, strength of parameter regularization, and dropout probability were simultaneously optimized to achieve optimal performance.
Architecture of HybridSucc
To maximally capture the sequence and structural properties of Ksucc sites, we combined the predictions of DNN and PLR, to create a deep-learning and conventional machinelearning architecture that can be called a hybrid-learning framework. For the general prediction of Ksucc sites, DNN was first used to train a computational model for each of the 10 types of features, and 4-, 6-, 8-, and 10-fold crossvalidations were performed to evaluate the robustness and accuracy of each model. For the 10 features (f1,f2,f3, ...,f10),one KSP(10,10)peptide was scored by DNN with 10 values(D1,D2,D3,...,D10).For the PLR,each KSP(10,10)peptide was given 10 scores(P1,P2,P3,...,P10)corresponding to the 10 features. Thus, each KSP (10, 10) peptide could be reencoded as a 20-dimensional number vector:
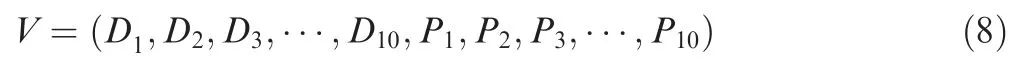
The vectorVwas used as the secondary feature and retrained by PLR to get a final score, whereas 4-, 6-, 8-, and 10-fold cross-validations were performed to evaluate the performance.For species-specific Ksucc models,transfer learning[31]was adopted by using the general models in DNN,and the species-specific data was used to fine-tune the network of each organism. All computational models were trained in a computer with an NVIDIA GeForce GTX 960 GPU, an Intel(R)CoreTMi7-6700K @ 4.00 GHz central processing unit (CPU),and 32 GB of RAM.
Prediction of potential Ksucc sites
First,we downloaded the complete proteome sequences of the 8 eukaryotes and 5 prokaryotes from UniProt (https://www.uniprot.org/,in December,2018).A classic approach of reciprocal best hits (RBHs) was adopted to determine potential orthologs of known Ksucc proteins in the 13 species, if two proteins of two different organisms reciprocally find each other as the best hit from the BLAST search. For each species, its corresponding predictor in HybridSucc was used to predict Ksucc sites in all known and orthologous proteins, with the default threshold (Sp= 95%). The prediction results were downloadable from http://hybridsucc.biocuckoo.org/download.php.
To identify potentially conserved and functional Ksucc sites, MUSCLE [32] (http://www.drive5.com/muscle/, version 3.8.31) was applied to perform multiple alignments for each group of orthologous proteins, and identify conserved Ksucc columns (CKCs) for aligned Lys residues that contained at least one known Ksucc site. For each CKC, organisms with a non-Lys residue, or a Lys residue that was not predicted to be a Ksucc site were removed.
Collection of human cancer mutations
We downloaded human somatic cancer mutations from the TCGA [24] data portal (http://portal.gdc.cancer.gov/, level 4 data, in May, 2018). All available projects were downloaded,including adrenocortical carcinoma (ACC), bladder urothelial carcinoma(BLCA),breast invasive carcinoma(BRCA),cervical and endocervical cancers (CESC), cholangiocarcinoma(CHOL),colon adenocarcinoma(COAD),lymphoid neoplasm diffuse large B-cell lymphoma(DLBC), esophageal carcinoma(ESCA), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), kidney chromophobe(KICH), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma(KIRP),acute myeloid leukemia(LAML), brain lower grade glioma (LGG), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), mesothelioma (MESO),ovarian serous cystadenocarcinoma (OV), pancreatic adenocarcinoma (PAAD), pheochromocytoma and paraganglioma(PCPG),prostate adenocarcinoma(PRAD),rectum adenocarcinoma(READ),sarcoma(SARC),skin cutaneous melanoma(SKCM), stomach adenocarcinoma (STAD), testicular germ cell tumors (TGCT), thyroid carcinoma (THCA), thymoma(THYM), uterine corpus endometrial carcinoma (UCEC),and uterine carcinosarcoma (UCS), and uveal melanoma(UVM). Entrez Gene IDs in TCGA files were used to map the mutation data to human Ksucc proteins. In total, we obtained 1,779,214 missense single nucleotide variants (SNVs)in 11,659 tumor samples across 33 major cancer types/subtypes.
A statistical approach for estimating functional impacts of KsuMs
In this study, we defined a KsuM as an SNV located within a KSP(10,10)region that potentially changes the protein Ksucc state.Since previous studies have demonstrated that the substitution of a Ksucc site to a glutamic acid (E) mimics the negatively charged succinyl group[12,33],a missense mutation of K to a non-E residue can directly disrupt a Ksucc site, and substitution of an SNV of a non-E residue to K might create a new Ksucc site. In addition,missense SNVs occurring at the flanking regions potentially increase or decrease the modification probability of a Ksucc site. To computationally identify KsuMs that significantly upregulate or downregulate protein Ksucc levels, we used the positive KSP (10,10) peptidesPand negative KSP (10,10) peptidesNto model the probability density distribution.For any given KSP(10,10)peptideKi,the scoreSicalculated by HybridSucc was transformed into a Bayesian posterior probability (BPP) as follows:
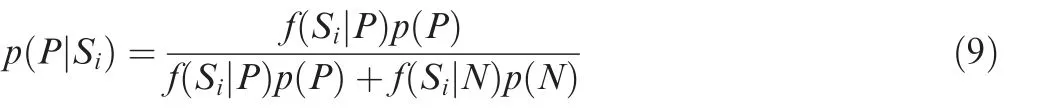
As previously described[34],the prior probability values ofp(P) andp(N) reflect our belief in the distribution ofPandNand were determined as the corresponding AUC value and 1,respectively. Then, HybridSucc was adopted to calculate the scores for all potential KsuMs (n= 63,693) inH. sapiensbefore (xi,i= 1, 2, 3, ...,n) and after (yi,i= 1, 2, 3, ...,n)the mutation, whereas all scores were normalized into BPPs.To estimate the global probability density distribution of BPPs before(x)and after(y)the mutation,we hypothesized that the joint probabilities within a small window might follow a normal distribution, and the Parzen window [35] based on the Gaussian kernel was used to conjugate the distributions in all windows to approximate the global distribution as follows:
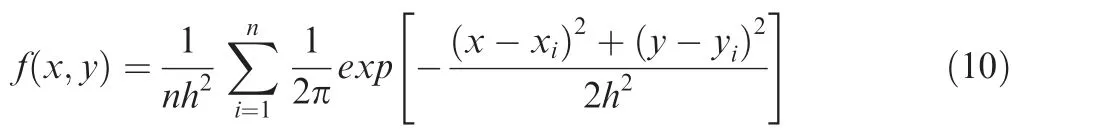
wherehis the window width,and the size ofhaffects the accuracy of the probability density estimation.The maximum likelihood estimation (MLE) method was used to determine the optimalhvalue as follow:
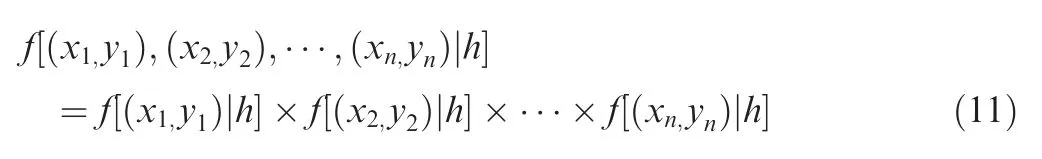
For differenthvalues(from 0 to 1,0.001 per step),we estimated the joint probability density distributions for each KsuM from the remaining ones, until all KsuMs were used once.The likelihood value is the product ofnprobability density values, and the optimalhvalue that maximized the likelihood value was determined to be 0.018 in this study. Finally,thexwas fixed, and the probability density distribution of its correspondingyvalues was computed. From the distribution, the statistical significance of a givenxscore was calculated with a threshold ofP<0.05. To prioritize potentially functional KsuMs, the mutated scoreyshould be >0.5 for KsuMs that potentially upregulate Ksucc levels, and the original scorexshould be >0.5 for KsuMs that potentially downregulate Ksucc levels. Only KsuMs that significantly influence known Ksucc sites were reserved.
Implementation of the web service
The online service of HybridSucc was constructed with PHP and JavaScript, in an easy-to-use manner with a user-friendly interface. The species can be selected and 3 threshold options including ‘‘High”, ‘‘Medium”, and ‘‘Low” can be chosen withSpvalues of ~95%,~90%,and ~85%,respectively(Table S8).We also implemented an ‘‘All” option to allow for the predictions on all Lys residues to be shown. HybridSucc was extensively tested on various web browsers including Internet Explorer, Mozilla Firefox, and Google Chrome to provide a robust and freely available service at http://hybridsucc.biocuckoo.org/.
Results
Ten types of sequence and structural features are efficient and informative
From public databases and the literature, we collected 26,243 non-redundant known Ksucc sites in 8830 proteins of 13 species.Our data set is much larger than those of previous studies such as pSuc-PseRat[36],in which 14,591 experimentally identified Ksucc sites were obtained in 4960 substrates (Figure 2A and B). Then, we eliminated homologous sites and identical peptides to prepare a benchmark data set, containing 21,770 non-homologous and non-identical KSP (10, 10) peptides in 7415 proteins for the general prediction (Figure 2C and D,Table S2). Such a procedure was also individually conducted to obtain 13 additional benchmark data sets for speciesspecific predictions. The results show that the species with the most abundant Ksucc sites in eukaryotes and prokaryotes areM.musculusandE.coli,which contained 6325 sites of 1639 proteins and 4161 sites of 1141 substrates, respectively (Figure 2C and D, Table S2).
Previously, 13 prediction tools were reported, while 10 types of sequence or structural features were established to encode Ksucc sites(Tables S1 and S3).The original PLR algorithm was improved by reserving the LASSO and adding two steps of random mutation and random zeroing, and then trained computational models for the 14 benchmark data sets,based on different types of features. The results show that sequence features were generally better than structural features(Figure 2E and Table S4).Due to the data limitation of protein structure databases,3 types of structural features were computationally derived from protein sequences in this work, and thus the predicted features might reduce the prediction accuracy. However, our results on different benchmark data sets showed AUC values >0.5, indicating that all features were efficient and informative for predicting Ksucc sites. For the general prediction, the AUC values of 10-fold crossvalidations ranged from 0.558 (ASA) to 0.729 (CKSAAP)(Figure 2E and Table S4). For species-specific predictions,the CKSAAP feature achieved the best performance inM.musculusandE. coli, with AUC values of 0.739 and 0.718,respectively. In contrast, our GPS feature outperformed other features in five organisms, includingR. norvegicus(0.801),S.cerevisiae(0.781),S. lycopersicum(0.761),B. subtilis(0.722),andC.glutamicum(0.761)(Figure 2E and Table S4).Thus,different types of features exhibited diverse accuracies for different data sets.
Moreover, we tested the 10 types of features by using two additional machine-learning algorithms, SVM and RF, and the results demonstrate that different algorithms also generated varying AUC values even for the same data set. For the general prediction, the GPS feature showed the best performance with AUC values of 0.668 and 0.741 for the SVM and RF algorithms,respectively(Table S4).The failure to find the most informative feature may be attributed to the intrinsic limitation of conventional machine-learning algorithms,which are less efficient on feature representation than deep-learning algorithms for large data sets [37]. Thus, we implemented a 4-layer DNN framework, including an input layer for inputting the data,two hidden layers for training the computational models, and an output layer for prediction. Unexpectedly,although the DNN-based results were generally better than PLR,SVM,and RF for species-specific predictions,four algorithms exhibited comparable results for the general prediction(Table S4).Thus,our results indicated that each type of feature only partially captured thebona fidecharacteristics of Ksucc sites, and no feature could achieve the best accuracy for all benchmark data sets.
Development of a hybrid-learning architecture for the prediction of Ksucc sites
Since all features were efficient and one did not outperform the others,we speculated whether the combination of all informative features can improve the accuracy. Also, since the DNNbased predictions did not show significantly better performance than PLR,SVM,or RF for individual features,we considered whether conventional machine-learning algorithms could be incorporated with the deep-learning algorithm to train a better model. Based on these two hypotheses, we designed a novel framework, HybridSucc, for the prediction of general or species-specific Ksucc sites from protein sequences (Figure 3A). For each benchmark data set, all positive and negative KSP(10,10)peptides were encoded by the 10 types of features, separately. Then, we used DNN and PLR algorithms to train a computational model for each feature,and 20 unique scores were outputted from the 20 models.Then the 20 scores were adopted as secondary features to be trained by PLR to obtain a single prediction value for each inputted KSP (10, 10) (Figure 3A).
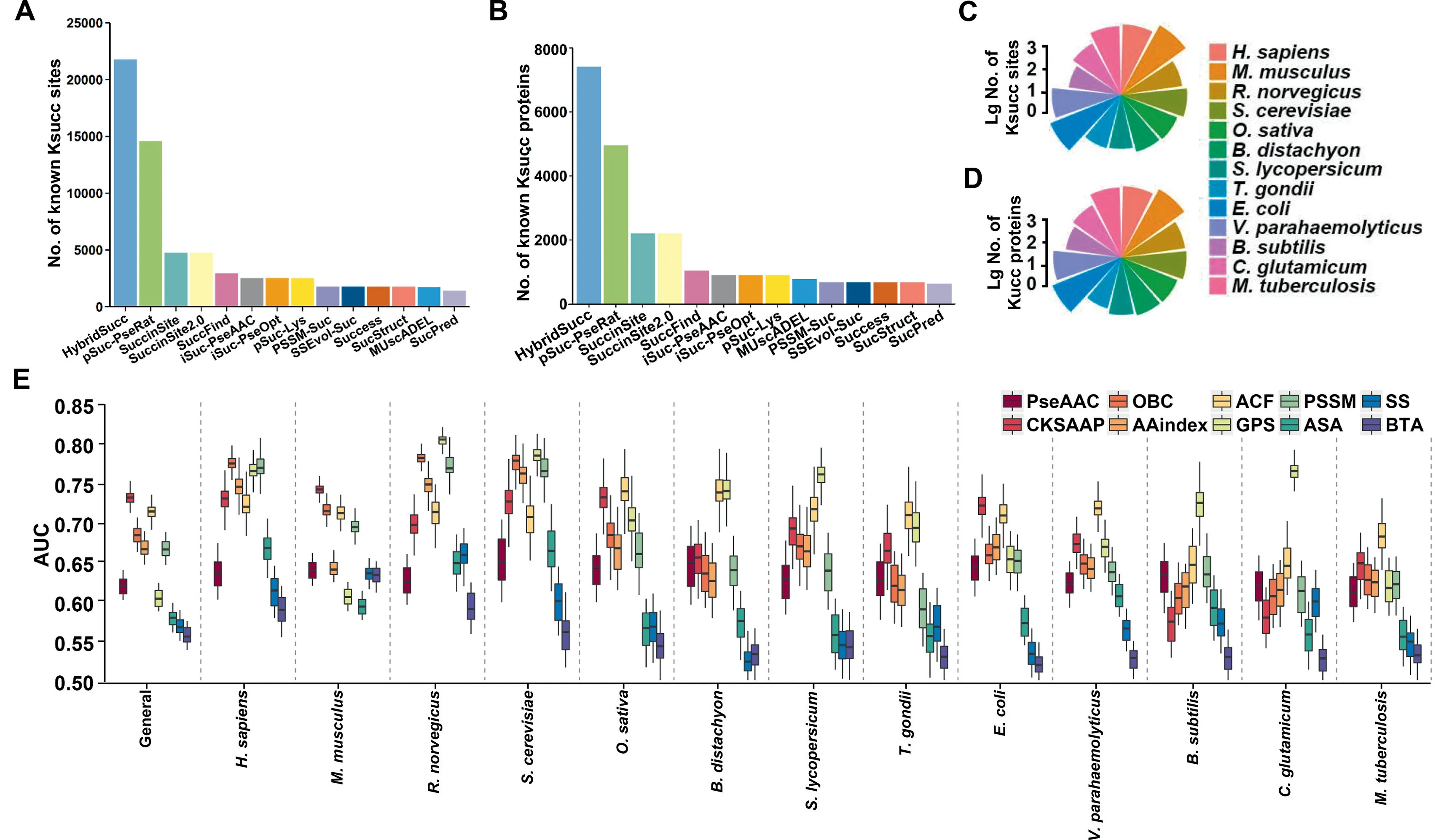
Figure 2 Benchmark data sets and prediction performance using different features
The 10-fold cross-validations showed that the AUC values of HybridSucc ranged from 0.840 to 0.961 (Figure 3B and C,Figure S1). Except the general prediction (0.885) and predictors in four prokaryotes,V. parahaemolyticus(0.887),B. subtilis(0.861),C. glutamicum(0.859), andM. tuberculosis(0.840), nine species achieved a highly satisfying performance with an AUC >0.9 (Figures 3B, C, and S1). For mammals,the AUC were 0.952, 0.920, and 0.961 inH. sapiens,M. musculus,R.norvegicus(Figures 3B,C,and S1).We also compared HybridSucc to single features individually trained by DNN or PLR algorithms and observed that the combination of the 10 types of features significantly improved the prediction performance for all benchmark data sets (Figure 3B). Furthermore,we adapted the HybridSucc architecture to only use the 10 scores generated from the DNN or PLR algorithm for a secondary training by PLR, and compared the results to those of HybridSucc (Figures 3C, S2, and S3). Exclusively using DNN or PLR generated AUC from 0.746 to 0.926, or 0.822 to 0.933, respectively, whereas HybridSucc achieved far superior accuracy on all benchmark data sets. For example, the DNN- and PLR-based AUC scores were only 0.819 and 0.878 inB. distachyon,respectively, whereas HybridSucc had an AUC of 0.920 (Figure 3C). Generally, HybridSucc exhibited a 2.05%–17.98% improvement of AUC values compared with individual algorithms (Figure 3C). Taken together, our results demonstrate that the combination of all informative features and the hybridization of conventional machinelearning and deep-learning algorithms could significantly improve the prediction performance for general and speciesspecific predictions.
Performance evaluation and comparison
To further evaluate the accuracy and robustness of Hybrid-Succ, 4-, 6-, and 8-fold cross-validations were also performed on the benchmark data sets. Due to the page limitations, the ROC curves ofn-fold cross-validations are shown for the general prediction(Figure 4A),and several species-specific predictors inH. sapiens(Figure 4B),O. sativa(Figure 4C), andS.cerevisiae(Figure 4D). The results show that the AUC of the 4-, 6-, 8-, and 10-fold cross-validations of HybridSucc for the general Ksucc data set were 0.875, 0.876, 0.882, and 0.885,respectively (Figure 4A). ForH. sapiens, the AUC values of 4-, 6-, 8-, and 10-fold cross-validations were 0.947, 0.950,0.952, and 0.952, respectively (Figure 4B), andn-fold crossvalidations also generated similar results inO. sativa(Figure 4C) andS. cerevisiae(Figure 4D). The high congruence of different cross-validation results indicates the promising accuracy of HybridSucc and that our computational models are robust.
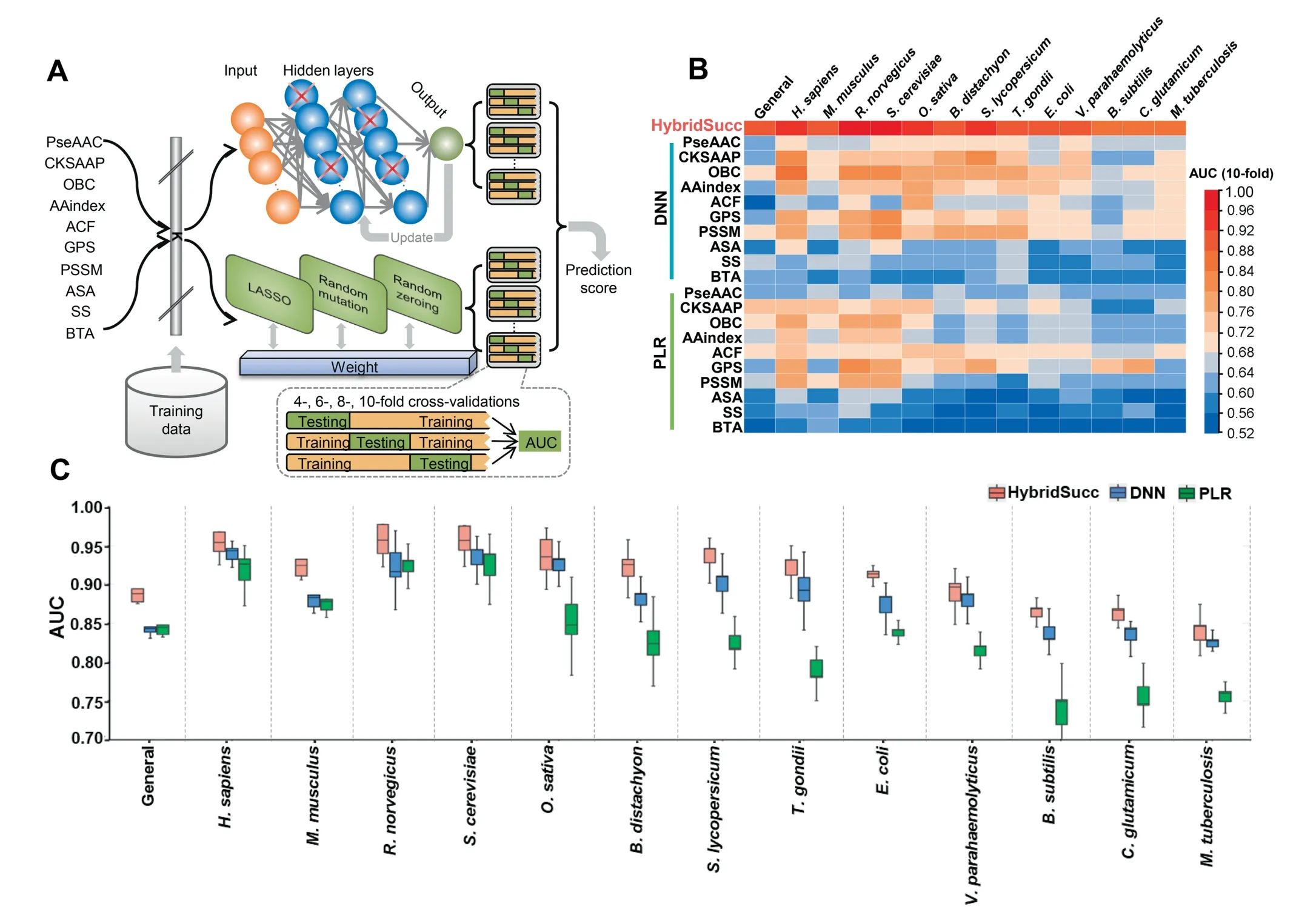
Figure 3 Hybrid-architecture and accuracy of HybridSucc
To demonstrate the superiority of HybridSucc, we compared the performance of HybridSucc with that of a number of previously reported and publicly available Ksucc site predictors, such as MUscADEL [19], SuccinSite [38], pSuc-PseRat[36], SuccFind [18], iSuc-PseAAC [21], pSuc-Lys [39], and iSuc-PseOpt[40](Table S1).Other methods without applicable online services were not included for the comparison. These 7 tools were developed for the general prediction and do not provide the option for customizing computational models from other data sets. Thus, we directly submitted the benchmark data set into each online service to calculate the performance and compare with the 10-fold cross-validation result of HybridSucc (Figure 4E). For MUscADEL [19], SuccinSite[38], pSuc-PseRat [36], and SuccFind [18] that output prediction scores for all Lys residues, ROC curves were illustrated and AUC values were computed as 0.751, 0.627, 0.700, and 0.742, respectively. For iSuc-PseAAC [21], pSuc-Lys [39],and iSuc-PseOpt [40], which have pre-defined cut-off values,we could only calculate the performance at the thresholds(Figure 4E). Compared with the second best tool MUscADEL[19],HybridSucc had a 17.84%higher AUC value(Figure 4E).For the species-specific prediction, we compared HybridSucc to SuccinSite2.0[20],which realized multi-predictors for seven species. We adopted the benchmark data set in SuccinSite2.0 as training data sets to retrain our models, and used the remaining positive and negative KSP (10, 10) peptides in our data sets as an independent testing data set. By comparison,HybridSucc outperformed SuccinSite2.0 [20] for all organisms examined (Figure 4F). Thus, HybridSucc implemented in the hybrid-learning architecture is significantly better than other existing tools.
Proteome-wide prediction of potential Ksucc sites
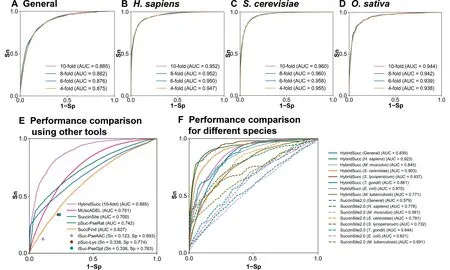
Figure 4 Performance evaluation and comparison of HybridSucc with other existing Ksucc prediction tools
First,potential orthologs of known Ksucc substrates in 13 species were computationally determined. Then we used Hybrid-Succ to conduct a stringent prediction (Sp = 95%) for both known and orthologous proteins (Figure 5A). We predicted 23,866 new Ksucc sites in 8710 proteins,while the species with the most predicted sites in eukaryotes and prokaryotes wereR.norvegicusandB. subtilis, respectively, which contained 4172 sites of 1298 proteins and 1417 sites of 552 substrates, respectively (Figure 5B and Table S2). To evaluate the reliability of the large-scale prediction, the amino acid occurrences around known and predicted Ksucc sites were analyzed and illustrated by pLogo[41](Figure 5C).The sequence preference of known Ksucc sites was highly similar to that of predicted ones. For example,there was an informative and over-represented acidic residue of aspartic acid (D) at the +1 position for both the known and predicted sites (Figure 5C). Thus, our predicted sites might be potential Ksucc sites with high confidence.
It has been demonstrated that a large proportion of PTM events might not be functional, and functional PTM sites evolve slowly[42].Thus,a Ksucc site evolutionarily conserved in more species might be functionally important with a higher probability. Based on this rationale, we multi-aligned protein sequences for each group of orthologous proteins. We obtained 3419 CKCs including 2624,436,and 359 CKCs containing 5251 known and 3615 predicted Ksucc sites conserved in eukaryotes, prokaryotes and both kingdoms, respectively(Figure 5D and Table S5). The distribution of the number of species for the CKCs showed that there were 49 CKCs with conserved Ksucc sites in ≥7 species (Figure 5D). The most conserved CKC was the aligned column of K301 in human heat shock protein family D(Hsp60)member 1(HSPD1),containing experimentally identified Ksucc sites in 10 species(Figure 5E).Human HSPD1 is a group I chaperonin that critically assists the correct folding of various mitochondrial proteins,and its active heptamers are formed from free monomeric molecules after being transported into mitochondria [43].Using HybridSucc, we also predicted that K301 of rat Hspd1 is a highly potential Ksucc site (Score = 0.8111), while this predicted site and known cognates might play a conserved role in regulating the self-assembly of HSPD1 (Figure 5E).
We also found that two CKCs contained known or potential Ksucc sites conserved in 10 species. One is K335 of the human succinate dehydrogenase complex flavoprotein subunit A(SDHA),which is a component of the SDH complex that is directly involved in the tricarboxylic acid cycle (TCA) cycle and electron transport chain [7] (Figure 5E). It has been reported that an increased Ksucc level of mouse SDHA promotes the SDH activity and cellular respiration [7], and such a function might be conserved for either known or predicted Ksucc sites in the aligned column. Another is K152 of human mitochondrial ribosomal protein S11(MRPS11),a member of mitochondrial ribosome [44]. Although only two Ksucc sites were experimentally detected in two prokaryotes,E. coliandV. parahaemolyticus, their orthologous sites in other species might also be succinylated and affect ribosome assembly.
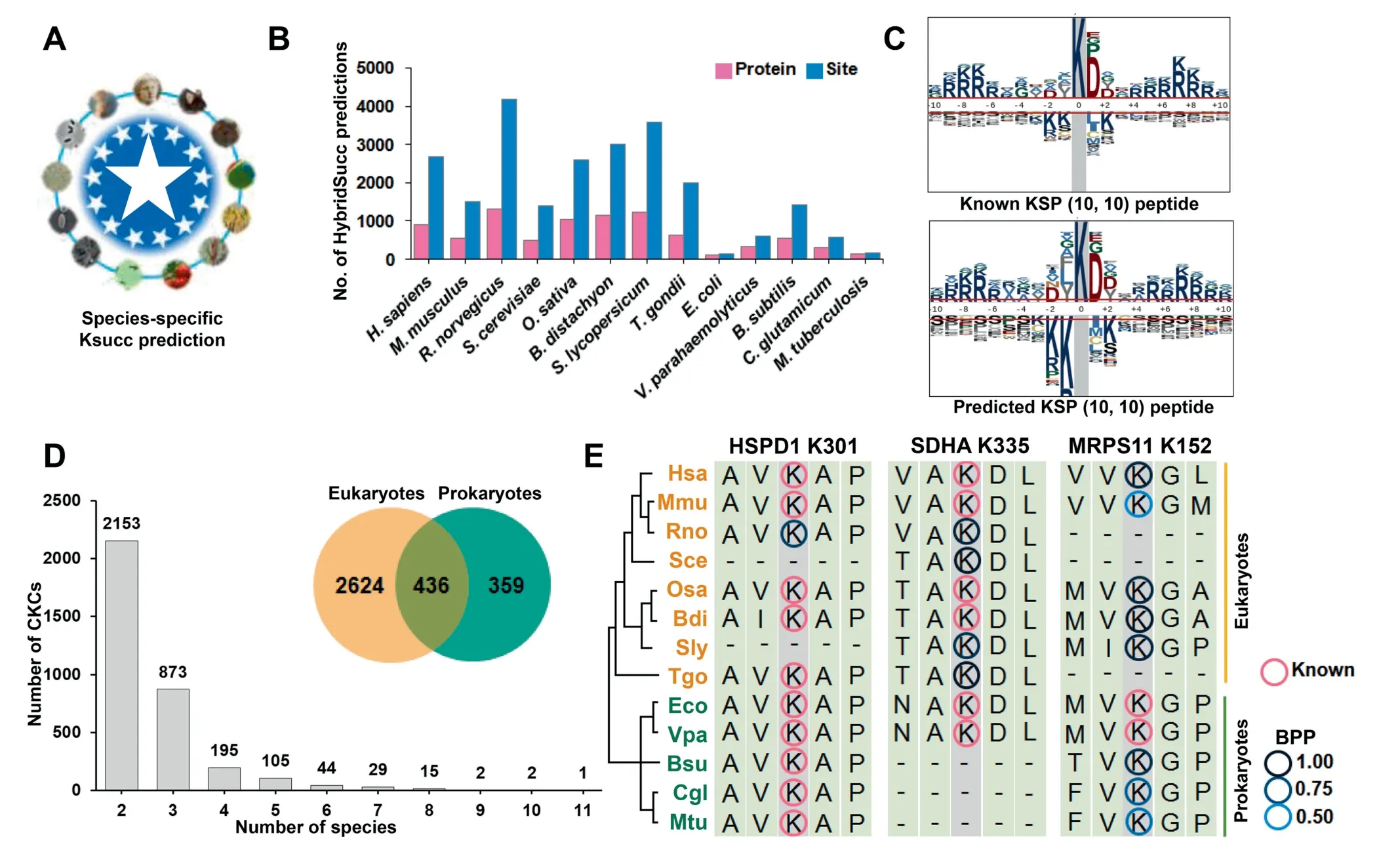
Figure 5 Proteome-wide prediction of Ksucc sites in 13 species
Prediction of potential cancer-associated KsuMs in H. sapiens
Cancer genome sequencing identified millions of missense SNVs in multiple types of human tumors [24], but exploring the functional impacts of these somatic mutations is challenging.Aberrant Ksucc is highly correlated with cancer[2,12],and a considerable number of missense SNVs might participate in tumorigenesis through changing protein Ksucc states. From 1,779,214 human cancer mutations, we detected 63,693(3.58%) potential KsuMs in KSP (10,10) regions. Then we developed a new statistical approach, GDPD, to prioritize 370 KsuMs in 218 genes that exhibited a significant impact on Ksucc sites(P<0.05;Figure 6A and Table S6).Moreover,we obtained 719 well-curated cancer genes from the Cancer Gene Census in the Catalogue of Somatic Mutations In Cancer(COSMIC)[45]and 2921 known human drug targets from the DrugBank database[46],and mapped the 218 genes to the two data sets. A hypergeometric test demonstrated that both the cancer genes and drug targets were significantly enriched in human KsuM-containing proteins, with enrichment ratios of 2.62-fold (P= 3.03E-04) and 4.15-fold (P= 1.20E-44),respectively (Figure 6B).
The results show a KsuM of R500Q from BRCA significantly decreased the Ksucc probability of PKM2 at K498(Figure 6C and Table S6), which is succinylated to increase the pyruvate kinase activity of PKM2 [25]. The desuccinylation of PKM2 K498 inhibits its activity to generate sufficient nicotinamide adenine dinucleotide phosphate (NADPH) to eliminate reactive oxygen species (ROS) and promote tumorigenesis, whereas the phosphomimetic substitution K498E significantly reduces cellular NADPH production and inhibits cell proliferation and tumor growth [25]. Therefore,we predicted R500Q as a deleterious KsuM that is potentially involved in the BRCA progression.We also observed a KsuM of R316W detected from UCEC that decreased the Ksucc of PKM2 at K311 (Figure 6C and Table S6), which is succinylated to inhibit the PKM2 activity [33]. Thus, R316W might have an effect opposite to that of R500Q and thus inhibit tumorigenesis.
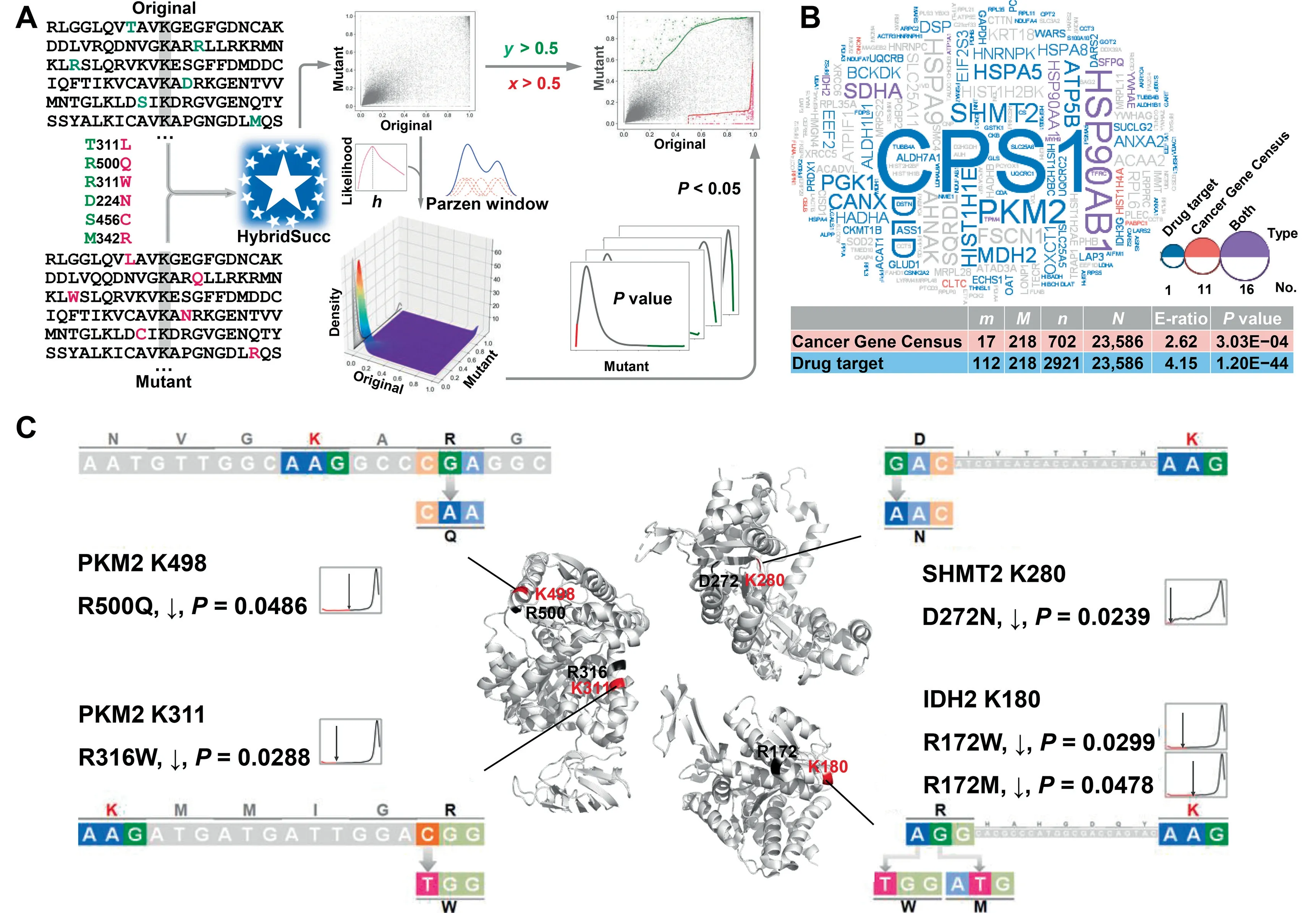
Figure 6 Prioritization of human KsuMs that significantly change protein Ksucc states in cancer
From SKCM and BRCA,a KsuM of D272N was predicted to significantly attenuate the Ksucc of K280 in SHMT2,a pyridoxal phosphate (PLP) binding protein (Figure 6C and Table S6). SHMT2 is activated through forming tetramers after binding PLP at the K280 site to promote tumor cell growth, whereas K280 Ksucc prevents PLP binding to decrease SHMT2 activity [12]. Thus, D272N might promote cancer progression through downregulating K280 Ksucc. In addition, the desuccinylation of IDH2, a NADPH-producing enzyme, enhances its activity, promotes the production of NADPH and protects cancer cells from oxidative damage[26]. Various R172 mutations activate IDH2 and frequently occur in human tumors such as glioma, LAML, and CHOL,although the exact mechanisms are unclear [47,48]. From LGG, we predicted two KsuMs, R172W and R172M, which might enhance the IDH2 activity by inhibiting the Ksucc level of K180 (Figure 6C and Table S6). In summary, our results indicated a strong correlation between Ksucc and human cancer, and prioritized 370 highly potential KsuMs for further experimental considerations.
Discussion
As a novel PTM [5,14], Ksucc plays a critical role in the regulation of numerous biological processes [6–11], and its dysregulation is highly associated with human diseases such as cancer[2,12].There has been rapid development in succinylomic profiling in the past few years, and thousands of Ksucc sites have been identified by high-throughput mass spectrometry.Due to the data accumulation, publicly available databases such as PLMD 3.0 [15], PhosphoSitePlus [16], and dbPTM [27] contribute to data sharing and reuse through the collection, integration, and annotation of known Ksucc substrates and sites. Based on these data resources, various algorithms and features were tested, and numerous computational tools wereconstructed to provide an alternative means for rapid identification of potential Ksucc sites in protein sequences[17,20–23].
In this study, we compiled a benchmark data set of 26,243 known Ksucc sites (Figure 2A and B), integrated 10 types of features, and merged DNN and PLR into a hybrid-learning architecture to develop a new tool, HybridSucc (Figure 3A).In the 10-fold cross-validation, HybridSucc showed AUC values of 0.885 and 0.952 for the general and human-specific predictions of Ksucc sites, respectively (Figure 4A and B), and achieved a ≥17.84%improvement of AUC values for the general prediction compared with other existing tools (Figure 4E and F). In order to explore correlations of different features represented by different algorithms, the 20-dimensional vectorV(D1,D2,D3, ...,D10,P1,P2,P3, ...,P10) initially scored by HybridSucc was retrieved for each KSP (10, 10) peptide around positive and negative sites in our benchmark data sets.The Kendall’s rank correlation was adopted to pairwisely measure relations between the 10 features. For the general prediction, average Kendall’s tau-b coefficients were calculated as 0.209 and 0.049 for DNN and PLR algorithms, respectively(Figure S4A and B),while the average correlation of captured features between DNN and PLR was determined as 0.081(Figure S4C). We also found a similar result for the humanspecific prediction, with average correlation values of 0.211,0.072, and 0.104 for DNN, PLR, and DNNvs. PLR, respectively (Figure S4D–F). Thus, our results indicate that these 10 types of features are independently and differentially represented by different algorithms. To further demonstrate the contribution of different features and algorithms to the final performance, we added one feature per time starting from BTA to retrain the DNN and PLR models, and calculated AUC values of 10-fold cross-validations for the general and human-specific predictions (Figure S5A and B). The gradual increase of AUC values indicates that all features and algorithms are crucial to enhance the prediction accuracy.
Although the mass spectrometry-based identification of thein vivosuccinylome has nearly become routine, such analyses are usually labor-intensive and expensive,and lowly expressed or succinylated proteins are difficult to probe. Thus, a largescale prediction of Ksucc sites from sequences can rapidly provide useful candidates for further experimental consideration.Using HybridSucc, a proteome-wide prediction of Ksucc sites was conducted for known substrates and their orthologous proteins in the 13 species.We predicted 23,866 potential Ksucc sites in 8710 proteins with a high stringency, and prioritized 5251 known and 3615 predicted Ksucc sites that are evolutionarily conserved with potentially important functions. From human cancer mutations, we identified 370 KsuMs in 218 genes that potentially change protein Ksucc states.The enrichment of cancer genes and drug targets in KsuM-containing genes indicates a strong correlation between Ksucc and human cancer.
In the future, we will continuously improve and maintain HybridSucc by collecting more experimentally identified Ksucc sites into the training data set.It should be noted that although the number of Ksucc sites used in HybridSucc is much larger than those in previous studies,a considerable number of Ksucc sites obtained from mass spectrometry-based identifications might be false positives.The development of methods for data quality control remains to be a great challenge to minimize the false positives generated by different types of experimental assays. In addition, we will test more useful features, and include more traditional or deep-learning algorithms into the framework. Taken together, this study reports a novel and accurate approach for the prediction of Ksucc sites.We anticipate that a hybrid learning architecture and the integration of multiple features can be easily extended to other types of PTMs to corroborate much better prediction.
Data availability
The benchmark data sets containing known Ksucc substrates and sites for general and species-specific predictions were prepared in the tab-delimited format,and UniProt accession numbers, protein sequences, Ksucc positions, organisms, and PubMed IDs (PMIDs) of original references were provided for Ksucc sites.Potential Ksucc sites predicted by HybridSucc were also accessible. All data sets and online service are freely available for academic use at http://hybridsucc.biocuckoo.org/download.php.
CRediT author statement
Wanshan Ning:Data curation, Formal analysis, Investigation,Methodology, Writing - original draft.Haodong Xu:Data curation, Formal analysis, Investigation, Methodology, Writing - original draft.Peiran Jiang:Data curation, Validation.Han Cheng:Data curation, Validation.Wankun Deng:Data curation, Validation.Yaping Guo:Data curation, Validation.Yu Xue:Conceptualization, Funding acquisition, Project administration, Writing - review & editing. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This study was supported by the Special Project on Precision Medicine under the National Key R&D Program of China(Grant Nos. 2017YFC0906600 and 2018YFC0910500), the National Natural Science Foundation of China (Grant Nos.31671360, 31801095, and 31601067), Fundamental Research Funds for the Central Universities (Grant Nos.2019kfyRCPY043 and 2017KFXKJC001), the National Program for Support of Top-Notch Young Professionals,Changjiang Scholars Program of China, program for HUST Academic Frontier Youth Team, and China Postdoctoral Science Foundation (Grant No. 2018M632870).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.11.010.
ORCID
0000-0002-1410-7891 (Ning, W)
0000-0003-2086-3893 (Xu, H)
0000-0003-1586-7806 (Jiang, P)
0000-0002-6684-317X (Cheng, H)
0000-0002-5052-9151 (Deng, W)
0000-0001-9937-363X (Guo, Y)
0000-0002-9403-6869 (Xue, Y)
 Genomics,Proteomics & Bioinformatics2020年2期
Genomics,Proteomics & Bioinformatics2020年2期
- Genomics,Proteomics & Bioinformatics的其它文章
- SuccSite: Incorporating Amino Acid Composition and Informative k-spaced Amino Acid Pairs to Identify Protein Succinylation Sites
- BGVD: An Integrated Database for Bovine Sequencing Variations and Selective Signatures
- SR4R: An Integrative SNP Resource for Genomic Breeding and Population Research in Rice
- IC4R-2.0: Rice Genome Reannotation Using Massive RNA-seq Data
- iGMDR: Integrated Pharmacogenetic Resource Guide to Cancer Therapy and Research
- MosaicBase: A Knowledgebase of Postzygotic Mosaic Variants in Noncancer Disease-related and Healthy Human Individuals