
Transcriptomic responses in resistant and susceptible maize infected with Fusarium graminearum
2020-04-19 02:30:24GuangshengYuanXiujingHeHuiLiKuiXiangLiLiuChaoyingZouHaijianLinJialiWuZhimingZhangGuangtangPan
The Crop Journal 2020年1期
Guangsheng Yuan*,Xiujing He, Hui Li, Kui Xiang, Li Liu, Chaoying Zou, Haijian Lin,Jiali Wu,Zhiming Zhang,Guangtang Pan
Key Laboratory of Biology and Genetic Improvement of Maize in Southwest Region of Ministry of Agriculture,Maize Research Institute,Sichuan Agricultural University,Chengdu 611130,Sichuan,China
Keywords:Gibberella ear rot Fusarium graminearum Molecular mechanism RNA-seq Differentially expressed genes
ABSTRACT Gibberella ear rot (GER) caused by Fusarium graminearum (teleomorph Gibberella zeae) is a common maize disease that not only severely reduces grain yield but also contaminates maize grain with mycotoxins. We investigated the molecular mechanism underlying the host defense responses against pathogen infection using comparative transcriptomic analysis.We injected F.graminearum spore suspensions into plants of resistant(IBM-81)and a susceptible (IBM-85) maize inbred line after pollination and performed RNA-seq 48, 72,and 96 h after inoculation. Respectively 487 and 410 differentially expressed genes (DEGs)were induced in the resistant and susceptible lines across three time points,indicating that a stronger defense response was activated in the resistant than in the susceptible line.Among them, 198 genes commonly induced in the two lines were subjected to pathway analysis, revealing that most of the DEGs were closely associated with defense and a wide range of metabolic activities.DEGs associated with pathogenesis-related protein 1(PR1)and regulation of salicylic acid were significantly enriched during F. graminearum infection,suggesting that these DEGs play dominant roles in maize resistance to GER. Our results provide a resource for future gene discovery and facilitate elucidation of the complex defense mechanisms involved in resistance to GER.
1. Introduction
The fungus Fusarium graminearum (Schwabe) is the primary pathogen causing Gibberella ear rot(GER)or stalk rot in maize[1,2].This pathogen is most common in cool and humid areas and is seen as a reddish-pink mold starting at the tip of a rotten ear [3]. Ingestion of F. graminearum-infected grain is harmful to both humans and livestock, owing to the production of diverse potent mycotoxins including deoxynivalenol(DON) and zearalenone (ZON) [4,5]. Chemical and agronomic methods preventing maize ear rot are often insufficient when climatic conditions are favorable for the pathogen. The most reliable and economical way to reduce the damage caused by ear rot would be to develop resistant cultivars[6].
Currently, there is no cultivar that is completely resistant to GER, given that the pathogenic races vary constantly with environmental influence [7]. Moreover, plant resistance against GER is under polygenic control. The use of markerassisted selection (MAS) to improve GER resistance is a promising strategy in resistance breeding programs [8]. The aim of MAS is to identify major resistance genes or quantitative trait loci(QTL)conferring high resistance to ear rot.Much progress has been made in understanding the resistance to GER caused by F. graminearum, including identification of disease resistance genes, detection of stable QTL, and characterization of defense responses [9-11]. Elucidating the molecular interaction between the fungus and the host will be hastened by isolation of resistance genes or QTL for maize ear rot, as well as by identifying the genetic mechanisms underlying defense response [12-14]. To date, few genes associated with GER resistance and limited QTL consistently expressed in diverse lines have been identified and their inheritance remains uncertain. In our previous studies[15-17], a suite of genes involved in resistance to ear rot were identified using a gene chip and 10 resistance QTL were mapped on various chromosomes, providing a valuable genetic resource for further functional genomic characterization of Fusarium ear rot resistance.
Most recently,advanced biotechnology has been deployed to identify new resistance genes,facilitating the investigation of molecular mechanisms of plant defense against fungal diseases [18,19]. For example, targeted chromosome-based cloning via long-range assembly, MutMap/MutMap+, resistance gene enrichment sequencing,and mutant chromosome flow sorting and sequencing (MutChromSeq) association genetics with resistance (R) gene enrichment sequencing have been used to dissect plant genotype-phenotype associations and clone resistance gene [2,20]. It is generally considered [19,21] that there are two layers involved in plant resistance to pathogen invasion: a first layer of pathogenassociated molecular pattern-triggered immunity (PTI) and a second layer of effector-triggered immunity(ETI).Infection of plants with diverse pathogens results in changes in concentrations of plant hormones including salicylic acid (SA),jasmonic acid (JA), and ethylene (ET), which have been established as playing crucial roles in the regulation of plant immune responses [22]. SA is generally involved in the activation of defense against biotrophic pathogens, while JA and ET are usually associated with defense against necrotrophic pathogens [23]. The host defense response to pathogen infection involves changes in the expression of large number of plant genes, leading to a reactive oxygen burst, signaling events, and a hypersensitive cell death response at the infection site.
Recently, large-scale approaches including transcriptional analysis and genome-wide association studies (GWAS) have been used to investigate the molecular basis of plant defense mechanisms, specifically for identifying effective QTL and candidate genes and resistance to mycotoxin accumulation[24-27]. Plants could respond to pathogen invasion through transcriptional regulation. Much progress has been made towards the understanding of defense processes of maize ear rot, and the RNA-seq approach has become a powerful technology for studying the interaction between host and pathogen at the transcriptional level. For instance, 81 genes were identified as contributing to resistance to GER using RNA-seq in maize [28]. Another study [29] investigated transcriptional alteration in response to Fusarium verticillioides and suggested that the small heat shock protein family,some secondary metabolites,and the signaling pathways of abscisic acid(ABA),JA,or SA may be involved in resistance to Fusarium ear rot(FER).A large number of differentially expressed genes(DEGs)and transcriptional changes were detected using RNAseq associating with maize Gibberella stalk rot (GSR), which two major locus conferring resistance to GSR through induced high expression of defense-related genes were described in the response of maize to F. graminearum infection [18].Although large number of genes involved in antifungal activities, signal transduction, secondary metabolite synthesis, and energy metabolism have been reported, it is difficult to determine which gene is involved in which biological process.
In this study, we investigated differential transcriptional regulation in a resistant and a susceptible maize inbred line at three different time points following F. graminearum infection,using RNA-seq. We confirmed representative induced genes involved in disease resistance using qRT-PCR.Indentified genes in both inbred lines after inoculation will pave the way towards better understanding of the molecular mechanisms underlying the resistance to maize ear rot disease caused by F.graminearum and provide valuable genetic information for further functional genomic studies in resistance to maize disease.
2. Materials and methods
2.1. Plant materials
Two maize inbred lines, the resistant IBM-81 and the susceptible IBM-85,were derived from the IBM Syn10 doubled haploid population (IBM Syn10 DH), which was developed from a cross between maize inbred lines B73 and Mo17 after 10 generations of intermating followed by DH line development [30]. The IBM Syn10 DH population are widely used in genetic studies for their high genetic resolution and phenotypic variation, and thus serve as an important resource for maize disease resistance research. Both IBM-81 and IBM-85 have been deeply resequenced across the whole genome,and carry similar genetic backgrounds [31] (Fig. S1). The genetic diversity present in the two inbred lines is a valuable resource for maize disease research (Fig. S2). IBM-81 and IBM-85 were subjected to preliminary evaluation for a three-year(2015-2017) field test by artificial inoculation with F.graminearum in southwestern China and were identified as showing respectively high resistance and susceptibility to GER. They were grown in the same growth chambers at the Agricultural Experiment Station of Sichuan Agricultural University,Chengdu,China,located at 30°33′40″N,103°39′3″E and each ear was enclosed separately in a kraft paper bag.
2.2. Inoculum production and disease inoculation
Spores of F. graminearum were cultured on potato dextrose agar medium for 15 days prior to collection for inoculations.Conidia were prepared by washing from the culture and dilution of the conidial suspension with sterile water.For field inoculation, the spore suspension was adjusted to a final concentration of approximately 1.0 × 106spores mL-1based on microscopic count. Inoculation was performed two weeks after pollination, by syringe injection of 3 mL of conidial suspension through the husk into the middle of the ear. At maturity, the proportion of infected area was estimated on a scale from 1 to 7, where grade 1 represents no visible disease symptoms and grades 2 to 7 describe visible symptoms on kernels with infection areas of 1%-3%, 4%-10%, 11%-25%,26%-50%, 51%-75%, and 76%-100%, respectively [32]. The severity score of each line was calculated as the mean score of 10 inoculated ears.
2.3. Sample collection
In our previous study [16], microscopic observation of pathogen invasion and physiological and biochemical assays showed that gene responses were induced gradually after 24 h post-inoculation (hpi) in maize Fusarium ear rot. We accordingly collected husk samples around the inoculation spots and from mock-inoculated plants at 48,72,and 96 hpi in an attempt to obtain highly enriched expression libraries.RNAs were extracted from husks of three ears, sampled from similar-sized areas around the inoculation spot, and mixed.Two independent biological replicates were assayed at each time point.
2.4. cDNA library preparation and sequencing
Total RNA was extracted using TRIzol (Invitrogen, Carlsbad,CA, USA) according to the manufacturer's instructions. The quality of RNA was verified using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). For RNA deep sequencing,approximately 6 μg of total RNA representing each library was used for library construction and Illumina sequencing.Briefly,mRNA was isolated and purified from total RNA using oligo(dT)magnetic beads and first-and second-strand cDNAs were synthesized. The cDNA was then ligated with adaptors at the 5′and 3′ends of the tags.The library was sequenced on an Illumina Hiseq PE150 platform. Clean reads were obtained by removing reads containing the adapter, reads containing poly-N and low-quality reads from raw reads. Correlations between gene expression levels in two replicates were calculated based on Spearman's correlation coefficients. The average proportions of clean reads in each sample were 95.66%-97.74%. The clean reads were used for further analysis.
2.5. Sequence analyses
The clean reads were aligned to the maize B73 reference genome(ZmB73_RefGen_V4)using TopHat 2.0.9 software[33].Only uniquely mapped reads (reads mapped to unique locations on the B73 genome) were retained for calculating gene expression and annotation. Alignments from each library were processed with Cufflinks 0.9.3 [34] to assemble transcript isoforms and quantify expression values,and then normalized as fragments per kilobase of exon model per million mapped reads (FPKM). To detect DEGs, statistical analysis among libraries was performed with a falsediscovery rate(FDR)threshold of 0.05 and a log2(fold change)using the DESeq package[35].To further assign and annotate the DEGs, Gene Ontology terms (http://www.geneontology.org/) and significantly enriched metabolic pathways in the Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.kegg.jp/)were identified using the genes in each library.
2.6. Quantitative real-time PCR analysis
To validate DEGs, twelve representative genes of different functional categories were subjected to quantitative realtime PCR (qRT-PCR) using the SYBR Premix Ex Taq protocol(TaKaRa Biotechnology, Dalian, Liaoning, China) on an Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems,Foster City,CA,USA)across three different time points for both maize inbred lines. The cDNA products were normalized for RT-PCR using 18S rRNA (forward primer: 5′-ATGTTCCGTGGCAAGATGAG-3′, reverse primer: 5′-CATTGTTGGGAATCCA CTC-3′) as an endogenous control.Specific primers used in sets for PCR were designed for target genes using Primer Premier 5.0 (http://www.premierbiosoft.com/index.html). Thermal cycle conditions were as follows:2 min at 95 °C followed by 40 cycles of 15 s at 95 °C, 15 s at 56-57 °C,and 15 s at 72 °C.Each PCR reaction was repeated at least three times.
3. Results
3.1. Phenotypic analysis of GER severity
Matured kernels of two inbred lines with contrasting GER symptom were photographed and assessed for disease severity. As shown in Fig. 1, the infected area in response to F. graminearum inoculation was smaller in the resistant line IBM-8 than in the susceptible line IBM-85,with a white or light pink mold observed on kernels.
3.2. Sequence assembly and mapping
Spearman's correlation coefficients were >0.8 for the two independent experiments and revealed both high correlations and slight variability among biological replicates,showing the reliability of the transcriptome sequencing data (Fig. S3).Between 33.16 and 46.38 million clean reads filtered from the raw reads of each library were aligned against the maize genome(B73 RefGen_v4).On average,86.28%of the total reads were mapped to the B73 reference genome, with most of the reads mapping to exon regions and only a small proportion mapping to introns or intergenic regions (Table 1). Incompleteness of the maize genome sequence data was probably one of the reasons for the occurrence of remaining unmapped reads.
3.3. Global analysis of differentially expressed genes

Fig.1-Phenotypic variation of GER severity in inbred lines IBM-81 and IBM-85 after F.graminearum inoculation.(A)Two ears of the resistant line IBM-81.(B)Two ears of the susceptible line IBM-85.The infected area is indicated by a red ellipse.
To identify dynamic changes at the transcriptional level in response to F.graminearum at three time points in both inbred lines,the DEGs were examined via comparison of expression levels in inoculated samples with those of their uninoculated controls using an FDR threshold of 0.05 and a log2(fold change) ≥1, and the transcript abundance of each gene was further calculated and analyzed as FPKM with Cufflinks [34].A total of 3956 DEGs were identified in the resistant line IBM-81 at the three stages (Fig. 2-A left). Of these 487 were common to all three stages,and 673,738 and 973 genes were found in the pairs T_48 h vs. T_72 h, T_72 h vs. T_96 h, and T_48 h vs.T_96 h,respectively.In the susceptible line IBM-85,5016 DEGs were identified at the three time points (Fig. 2-A right). Of these, 410 DEGs were common to all three stages,and 1010, 571 and 619 genes were uniquely expressed in the pairs T_48 h vs.T_72 h,T_72 h vs.T_96 h,and T_48 h vs.T_96 h, respectively. Comparison of the DEGs in each library across the two inbred lines showed that the number of DEGs were decreased quickly as time went on in the susceptible line, indicating the variation tendency of the transcript levels in IBM-81 was more stable than that in IBM-85. This result suggests that the DEGs involved in the F. graminearum infection were differently regulated in the resistant and susceptible inbred lines.
Based on common DEGs to all three stages from each inbred line, further analysis of the conserved DEGs wasperformed. A total of 466 DEGs with consistent up- or downregulation and 21 DEGs with irregular expression across three time points were identified in IBM-81. Similarly, 379 DEGs with consistent regulation and 31 irregular DEGs across the three points were identified in IBM-85 (Fig. 2-B). Thus, the number of specific DEGs in the resistant line was greater than that in the susceptible line, possibly accounting for the different resistance levels in the two lines. Overall, 198 DEGs were common to the two inbred lines, suggesting that these genes should be focused on their activities and probably involved in the response to F. graminearum infection. The expression profiles of common DEGs were clustered into five groups, of which all but the fourth were up-regulated,suggesting that the abundance of expression levers of DEGs involved in defense might be greatly increased during F.graminearum invasion(Fig.2-C).
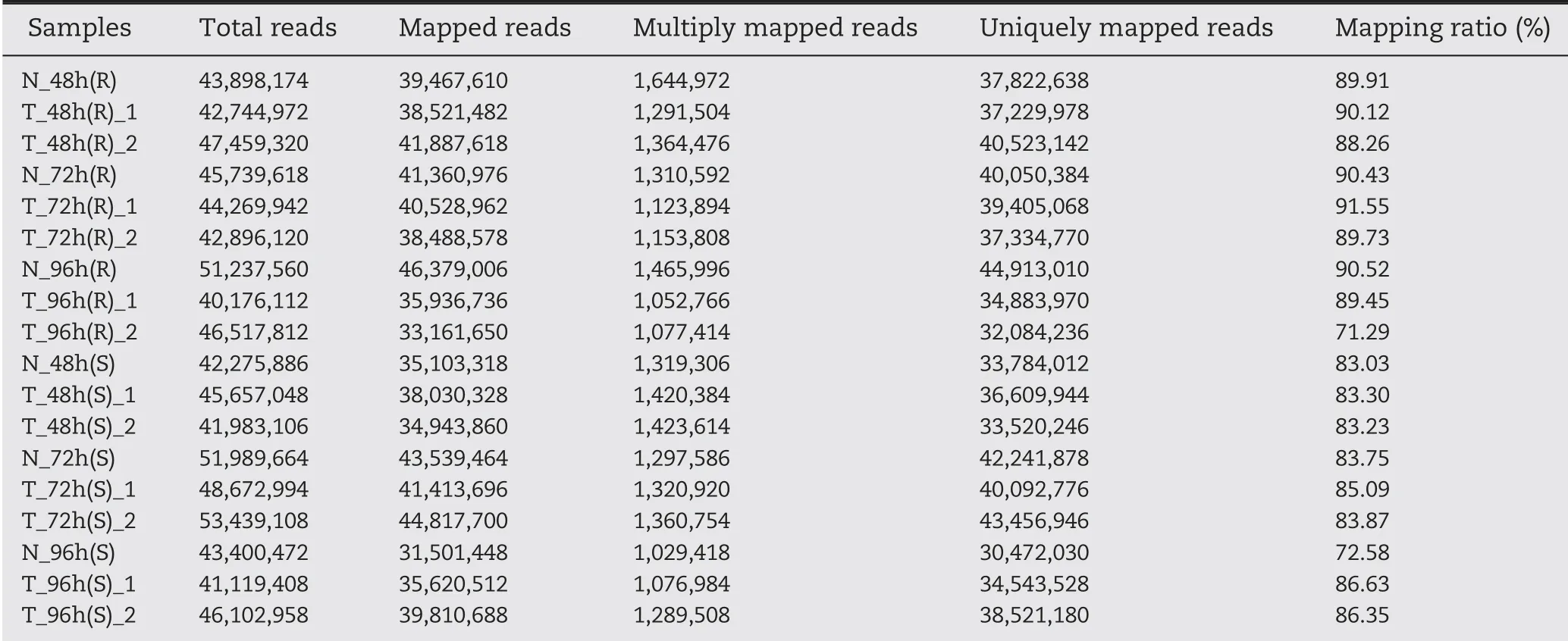
Table 1-Clean reads mapped to the maize B73 genome.
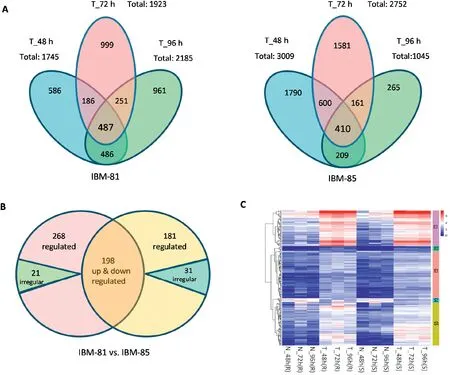
Fig.2-Genes differentially expressed at three time points in two maize inbred lines.(A)Venn diagrams of DEGs in inoculated samples of the resistant line IBM-81 and the susceptible line IBM-85(vs.non-inoculated samples).Overlaps show the number of genes commonly differentially expressed at the three time points.(B)Venn diagrams of genes expressed in common in the two inbred lines.Up,down,and irregular:genes up-or down-regulated or irregularly expressed.Overlaps show the numbers of genes shared between the two inbred lines.(C)Clustering and heat map of DEG expression levels in the two inbred lines.The color scale indicates the fold changes of gene expression.Sample names are displayed below the heat maps.A fold change of≥2 is shown in red(increased transcript abundance),a fold change of ≤2 is shown in blue(decreased transcript abundance),and no change is shown in white.DEGs were classed into G1,G2, G3,G4, and G5 according to their transcript abundance, and indicated with groups 1,2,3,4,and 5.
3.4. Pathway enrichment analysis of specific DEGs in both inbred lines
Pathway enrichment analysis was performed to investigate the biological pathways associated with the DEGs unique to each inbred line and the 198 DEGs common to the two lines.Enriched representative Gene Ontology (GO) terms of specific DEGs revealed that a large proportion of genes were strongly associated with plant disease resistance pathways (Fig. 3-A),such as defense response (GO:0006952), response to salicylic acid (GO:0009751), and protein kinase activity (GO:0004672).Because the transcripts abundance of specific DEGs associated with defense response were significant, they could be considered as a distinct mechanism of defense in different inbred lines. Interestingly, the term “response to salicylic acid”(GO:0009751)was highly represented in IBM-81,suggesting an important role of SA pathways in the defense resistance to F. graminearum infection.
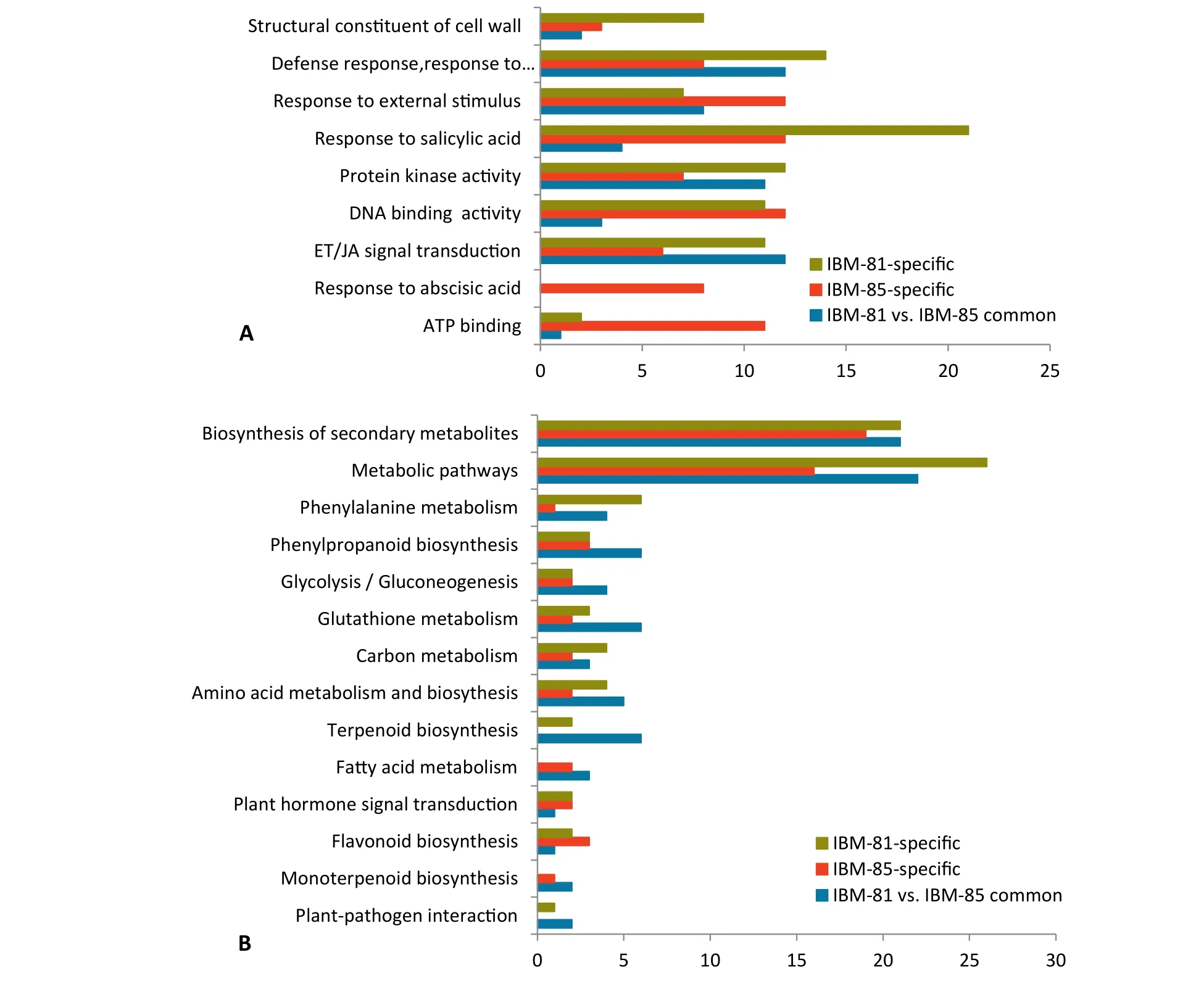
Fig.3-Functional analysis of specifically regulated genes after inoculation.(A)Enriched GO terms associated with specifically regulated genes in two inbred lines after F.graminearum inoculation.The ordinate represents major biological process GO terms.The abscissa represents the numbers of genes associated with each process.(B) Enriched KEGG pathways associated with specific genes in the two lines.The ordinate represents major biological pathways in KEGG.The abscissa represents the numbers of genes associated with each pathway.
To identify the biological functions of specific DEGs,KEGG pathway analysis was performed with a corrected Pvalue ≤ 0.05. Several KEGG pathways were significantly enriched after F. graminearum infection, including synthesis of secondary metabolites, metabolic pathway, carbon metabolism, glycolysis/gluconeogenesis, glutathione metabolism,phenylpropanoid biosynthesis, and phenylalanine metabolism(Fig.3-B).Among these pathways,synthesis of secondary metabolites and metabolic pathway were enriched more highly than the others, indicating the involvement of more genes participating in synthetic and metabolic processes in response to F. graminearum invasion.
3.5.Characteristics of genes associated with disease resistance among DEGs
Our primary objective was to identify genes associated with disease resistance and undergoing dynamic change at the transcriptional level during F. graminearum infection. We also queried the putative functions of DEGs based on information from Ensembl Plants (https://plants.ensembl.org/) and the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/) database. Numerous DEGs were identified as being involved in disease resistance processes.For instance, genes encoding disease resistance protein(Zm00001d014650, Zm00001d014649, and Zm00001d006755),as well as other pathogenesis-related protein genes(Zm00001d018738, Zm00001d011737, and Zm00001d048950),involved in direct response to pathogen invasion were identified. Genes encoding protein kinase (Zm00001d018502, Zm00001d017613,Zm00001d006536,and Zm00001d037397),ERF protein (Zm00001d027925), glutathione transferase 23(Zm00001d020780), and chitinase 1 (Zm00001d032947) were induced after F. graminearum infection, and thus led to hypersensitivity, regulated defense-related genes and activated resistant capability.
Twelve genes containing typical domains of the pathogenesis-related protein 1 (PR1) family were identified,and may act by enhancing typical resistance activities in defense systems (Fig. 4). Based on the phylogenetic relationship of these gene families, these 12 genes showed a high degree of similarity in their protein motifs, and may encode the same PR1 protein. The expressions of four of the genes were significantly altered between the treatments and control, with Zm00001d029558 more highly expressed in IBM-81 than in IBM-85 and the expression of Zm00001d009296,
Zm00001d018734, and Zm00001d018738 increasing gradually following F. graminearum infection. The GO and KEGG annotation showed that these four genes were involved in MAPK signaling pathway,salicylic acid-mediated signaling pathway,and biological process of plant-pathogen interaction, which have been proposed to be strongly associated with plant disease resistance [22].
3.6. Validation of candidate gene expression
To validate the results obtained by RNA-seq, we performed a parallel qRT-PCR analysis describing expression patterns with log2(fold values).Twelve representative genes:Zm00001d003190(chitinase A1), Zm00001d014649 (disease resistance protein RPM1), Zm00001d017139 (hydroxymethylbutenyl diphosphate synthase1),Zm00001d018734(PR1),Zm00001d020780(glutathione transferase), Zm00001d028815 (PR 10), Zm00001d033455 (Protein P21), Zm00001d042143 (glucosidase), Zm00001d048950, and Zm00001d048948 (hevein-like preproprotein), Zm00001d051001(glyceraldehyde-3-phosphate dehydrogenase) and Zm00001d053952(bax inhibitor 1),which are involved in response to pathogen infection and defensive pathways,were selected for further analysis(Fig.5).The expression levels of these representative genes relative to control as measured by qRT-PCR showed good agreement with those measured by RNA-seq.Zm00001d003190, associated with chitinase activity, displayed different expression patterns in the two lines,being up-regulated in the resistant and down-regulated in the susceptible line.Although expression of a few genes showed slight variation relative to their RNA-seq expression,there was a strong positive correlation between qRT-PCR and RNA-seq expression levels.
4. Discussion
4.1. Superiority in distinct phenotypic trait and RNA sequencing
In maize,the IBM Syn10 DH was obtained after 10 generations of F2intermating (B73 × Mo17 hybridization) and showed a diversity of phenotypic traits[31].Both lines IBM-81 and IBM-85 were picked out for their typical GER symptom, and the smaller infected area in the inbred line IBM-81indicates that certain defense mechanisms were probably activated in resistant line. High-throughput RNA sequencing has been reported as an effective approach to identifying resistance genes at the transcription level and is widely used in previous studies of maize resistance to disease[36].Based on RNA-seq,1255 causal transcripts were identified following F.graminearum infection in GER [28], a large number of responsive genes were identified following F.verticillioides invasion in FER[29],and thousands of induced genes were detected after F. graminearum inoculation in GSR [18]. In the present study,
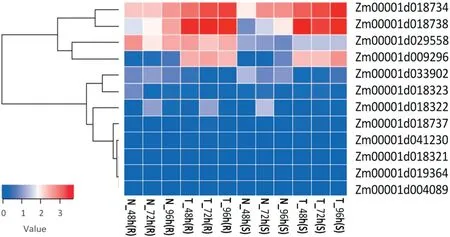
Fig.4-Clustering and heat map of PR1-family expression levels at three time points in two maize inbred lines.The color scale indicates the fold changes of gene expression.Sample names are displayed below the heat map.A fold change of ≥2 is shown in red(increased abundance),a fold change of ≤2 is shown in blue(decreased abundance),and no change is indicated in white.
we also identified a large number of DEGs during F.graminearum infection in two inbred lines by the highthroughput method(Fig.2-A).Expression of genes specifically in the current study indicated that the interaction between host and pathogen results in induction of a range of genes playing different roles in plant defense, as supported by the qRT-PCR validation of 12 representative genes on the basis of RNA-seq expression(Fig.5).
4.2. Regulation of specific pathways response to F.graminearum infection
After perception of a pathogen infection, defense responses of a host to pathogen infection are activated through a complex network to orchestrate the establishment of defensive barriers against the pathogens, which includes regulation of defense gene expression, induction of programmed cell death, defense response to fungus, metabolism and synthesis pathways, and SA, JA, and ET signaling pathways[9,19,23]. In the present study, several genes involved in SA signaling pathways were particularly responsive to F.graminearum and could be considered signal regulators coordinating the differential expression of many gene or protein complexes to participate in defense processes(Fig.3-A). In support of this observation, in previous studies, genes associated with SA signaling pathways were significantly induced in Fusarium-treated samples[28,29].Based on KEGG,we found that synthesis of secondary metabolites and metabolic pathway were significantly enriched in response to F.graminearum,clearly suggesting that both pathways play essential roles in activating or repressing transcription in maize resistance to fungal infection (Fig. 3-B). This strong transcriptional response in pathogen-caused disease has also been reported previously, with genes involved in metabolism and secondary metabolite biosynthesis pathways found to be responsive to maize ear rot [18,19,28,29].We also found that a series of DEGs associated with defense activities were induced after F. graminearum infection.For instance, genes encoding pathogenesis-related protein(Zm00001d018738, Zm00001d032947, and Zm00001d048950),protein kinase (Zm00001d018502, Zm00001d037397,Zm00001d006536, and Zm00001d017613), disease resistance RPP13-like protein (Zm00001d006755), NBS-type resistance activity (Zm00001d011737), and response secreted protease(Zm00001d019450) were differentially expressed following F.graminearum infection,suggesting that these genes may play direct or indirect roles in antifungal activities during pathogen invasion. Likewise in previous studies, several genes encoding PR proteins showed high expression in the resistant inbred line CO441 during F.graminearum infection in GER[28],and protein kinases were quickly induced to activate the PTI defense responses after F. verticillioides invasion in FER disease [29]. The disease resistance protein Zm00001d006755 was induced in resistance to gray leaf spot disease[37].All of these named genes or proteins were identified as being involved in response to F. graminearum infection and may play essential roles in resistance to fungal infection in maize.
4.3. PR1 family play crucial role in resistance to GER disease
PR1 proteins consist of 17 subfamilies [38] and have been often considered as markers for the enhanced defensive state conferred by pathogen-induced systemic acquired resistance(SAR)and have been reported extensively for their antifungal activities [39]. Interestingly, 12 genes containing typical PR1 family domains were specifically induced in response to F.graminearum inoculation in our study (Fig. 4). The expression levels of Zm00001d018734, Zm00001d018738, Zm00001d029558,and Zm00001d009296 were greatly changed between treated and control samples, with the expression of Zm00001d029558 higher in IBM-81 than in the susceptible line IBM-85. The expression level of Zm00001d018734 was dramatically increased in the inoculated sample compared to the control.ZmPRms (Zm00001d009296) has been speculated [40] to be involved in resistance to infection by Aspergillus flavus in maize and to play a key role in defense. The PR protein Zm00001d018738 was reported[41]to be involved in the MAPK signaling pathway and to be a member of stress-related resistance-associated proteins (RAPS), which play critical roles in host defense to Aspergillus flavus infection in maize.The induced genes Zm00001d029558 and Zm00001d018734 were annotated as PR1 proteins and probably participate in GER disease. These results clearly suggest that specific PR1 families were highly induced in response to F. graminearum infection,implying their crucial effect on GER resistance.
4.4. Induced DEGs involved in resistance processes in GER disease
We performed a comprehensive transcriptome analysis and identification of DEGs during F.graminearum inoculation using RNA-seq.A total of 3956 and 5016 transcripts were induced in the resistant inbred line IBM-81 and susceptible one IBM-85 across three time points, in comparison with controls, and large numbers of genes were identified as being associated with plant defense responses and key pathways during pathogen attack (Fig. 3). We also found that representative pathways involved in synthesis of secondary metabolites,metabolic pathways, glycolysis-associated pathways, glutathione metabolic pathways, phenylpropanoid biosynthesis,and phenylalanine metabolism were enriched in the plantpathogen interaction, indicating that the pattern of PTIinduced or ETI-induced acquired systemic immunity was initiated in maize against F.graminearum invasion.In support of this observation are similar results from previous studies,in which the processes of terpenoid biosynthesis (KO:00900)and phenylpropanoid biosynthesis (KO:00940) were enriched in response to Fusarium-caused ear rot in maize [29,42]. It is well known[43,44]that hormone signaling plays crucial roles in response to biotic and abiotic stress in plants. In the present study, the transcriptional abundance of SA pathway genes was dramatically increased in IBM-81 in response to F.graminearum infection, suggesting a major biological process of SA-mediated signaling pathways in resistance to maize GER. Further extensive studies are required to address the detailed molecular mechanisms underlying mediation by these genes of GER resistance in maize.
5. Conclusions
As a first step towards the identification of genes differentially induced at the transcriptomic level in maize by F.graminearum attack, RNA-seq has been shown to be a particularly suitable approach for identifying candidate genes involved in disease defense.By comparing the responses of resistant and susceptible inbred lines to F.graminearum infection,we identified 897 specific DEGs with possible functions in defense against F.graminearum infection. In particular, PR1 families and the SA pathway seem to play essential roles as regulators during maize defense response to GER.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31471513, 31601316) and Innovation Training Program of Sichuan Agricultural University(201710626050). We are very grateful to Prof. Xiquan Gao(Nanjing Agricultural University, China) for critically reading this manuscript.
Appendix A.Supplementary data
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2019.05.008.

- The Crop Journal的其它文章
- Brief Guide for Authors
- Rapid generation advance(RGA)in chickpea to produce up to seven generations per year and enable speed breeding
- Relay-intercropping soybean with maize maintains soil fertility and increases nitrogen recovery efficiency by reducing nitrogen input
- Genetic analysis and QTL mapping of stalk cell wall components and digestibility in maize recombinant inbred lines from B73 × By804
- Genetic bases of source-, sink-, and yield-related traits revealed by genome-wide association study in Xian rice
- Performance and yield stability of maize hybrids in stress-prone environments in eastern Africa