
Genetic analysis and QTL mapping of stalk cell wall components and digestibility in maize recombinant inbred lines from B73 × By804
2020-04-19 02:30:14QiWangKunLiXiaojiaoHuHuiminShiZhifangLiuYujinWuHongwuWangChanglingHuang
The Crop Journal 2020年1期
Qi Wang, Kun Li, Xiaojiao Hu, Huimin Shi, Zhifang Liu, Yujin Wu,Hongwu Wang,*, Changling Huang,*
aNational Key Facility for Crop Gene Resources and Genetic Improvement,Institute of Crop Sciences,Chinese Academy of Agricultural Sciences,Beijing,China
bNational Engineering Laboratory for Crop Molecular Breeding,Beijing,China
Keywords:Maize QTL Cell wall Digestibility Recombinant inbred lines
ABSTRACT The cell wall composition and structure of the maize stalk directly affects its digestibility and in turn its feed value.Previous studies of stem quality have focused mostly on common maize germplasm, and few studies have focused on high-oil cultivars with high grain and straw quality.Investigation of the genetic basis of cell wall composition and digestibility of maize stalk using high-oil maize is desirable for improving maize forage quality. In the present study, a high-oil inbred line (By804) was crossed as male parent with the maize inbred line B73 to construct a population of 188 recombinant inbred lines (RILs). The phenotypes of six cell-wall-related traits were recorded, and QTL analysis was performed with a genetic map constructed with SNP markers. All traits were significantly correlated with one another and showed high broad-sense heritability. Of 20 QTLs mapped, the QTL associated with each trait explained 10.0%-41.1% of phenotypic variation. Approximately half of the QTL each explained over 10%of the phenotypic variation.These results provide a theoretical basis for improving maize forage quality by marker-assisted selection.
1. Introduction
The maize stalk plays a critical role in plant growth,providing the main support for the plant, and it is the main forage resource for ruminants[1].
The stem cell wall is a high-fiber complex that encloses the cytoplasm and plays a major role in maize growth and development [2]. It is composed mainly of lignin, cellulose,and hemicellulose, which are also the major components of stem dry matter. There are four indices for the evaluation of stalk forage quality: acid detergent fiber (ADF), neutral detergent fiber (NDF), acid detergent lignin (ADL), and digestibility. ADF and NDF quantify fiber and ADL quantifies lignin[3,4].Because determining digestibility in vivo is difficult and its results are complex and non-intuitive, researchers have proposed that in vitro is highly correlated with in vivo digestibility,including in vitro dry matter digestibility(IVDMD)and in vitro neutral detergent fiber digestibility (IVNDFD).However, it is laborious and expensive to quantify cell wall components and IVDMD by chemical methods. In recent years,near-infrared reflectance spectroscopy(NIRS),which is low-cost, high-speed, and highly reproducible, has been widely used to evaluate the fiber content and degradation of maize stem cell walls[5].
Degradation of the maize stalk is negatively correlated with ADF,NDF,and lignin and is the key factor for improving forage quality [6]. Fiber and especially lignin are the main components limiting stem digestion. Reducing the content and concentration of fiber and lignin will increase the digestibility of cell wall and even dry matter, thereby increasing the feed value of maize [3,7]. To achieve this goal,geneticists have made efforts to identify many of the genes for enzymes involved in lignin biosynthesis and to regulate the expression of genes related to lignin synthesis by molecular biological means including overexpression, antisense RNA,and RNAi[3,8,9].Six brown midrib(bm)mutants of maize have been developed and are excellent materials for improving maize forage quality because of their low lignin content and/or changes in their lignin composition that increase cell wall digestibility.However,the application of bm for reducing stalk lodging resistance and increasing growth rate, and grain and dry matter yield has been minimal,especially for bm3[10,11].It seems impossible to improve feed value by molecular manipulation or use of mutants, and conventional breeding technology that relies on phenotypic selection is timeconsuming and offers low accuracy. With the development of molecular genetics and molecular quantitative genetics,marker-assisted selection has become an important method for breeding new varieties,which could speed up the breeding process. Identifying quantitative trait loci (QTL) for cell-wallrelated traits and characterizing their distribution and effects will provide a theoretical basis for the fine-mapping and mapbased cloning of QTL and improvement of maize silage quality.
From 1995 to 2010, fourteen QTL mapping studies were performed for cell wall components and digestibility in various populations [12]. Regions considered to be hotpots of QTL for cell-wall-related traits are bins 1.11, 2.08, 3.05, 5.04,6.05, 8.03, 9.04, and 9.06 [12]. However, these studies rarely employed high-oil maize and were performed mainly with low-density restriction fragment length polymorphism(RFLP)or simple sequence repeat (SSR) markers. The results of previous studies of 50 normal maize inbred lines and 50 high-oil maize (HOM) inbred lines (including By804) showed that the stalk quality of HOM was higher than that of normal maize [1]. So it is possible to breed silage maize hybrids with excellent forage digestibility and high biological production by crossing these two kinds of inbred lines. Thus, it is very important to make efforts to study the genetic characteristics of cell-wall-related traits of HOM stalks for maize forage breeding with single-nucleotide polymorphism (SNP)markers, which have the advantages of high density, high throughput, stability, and widespread use in plant genetic linkage map construction, linkage disequilibrium analysis,and QTL mapping[13].
Cell wall component accumulation is influenced by population and environment and changes continuously during plant growth and development [14]. The silking stage is the critical period of energy accumulation in the life of maize.Cellulose and hemicellulose contents reach their maximum by this stage [14].The objectives of the present study were to dissect the genotypic and environmental effects on stalk cell wall components and digestibility, identify correlations between them, and identify QTL associated with cell-wallrelated traits at the silking stage.
2. Materials and methods
2.1. Plant materials and field experiments
A population of 200 RILs was developed from a cross between maize inbred lines B73 and By804. B73 is an elite inbred line with low to intermediate levels of cell wall components and By804 is a high-oil inbred line from China Agricultural University. The hybrid F1was self-pollinated to produce an F2population that was then advanced to the F10generation by single-seed descent (SSD). Owing to pollen contamination during population development, 12 RILs with high (>10%)heterozygosity were excluded for further analysis,leaving 188 RILs.
The 188 RILs and parents were planted at Hainan in 2012 and Beijing in 2013 in a randomized complete block design.Each line was grown with three replicates in a single 5.5 m row, with a row spacing of 0.6 m and a planting density of 60,000 plants ha-1.
2.2. Phenotype evaluation
At the silking stage, three plants with similar growth were selected from each row and the stem from the second to fourth internodes from the ground was collected from each.Then samples were immediately placed in an oven at 105 °C for 30 min to inactivate intracellular cellulase and dried at 65 °C. The dried stem samples were comminuted with a hammer crusher and sieved through 0.5-mm mesh. The powdered samples were further dried at 45 °C for 48 h.Cellulose (CEL), lignin (ADL), ADF, NDF, and IVDMD were evaluated with a near infrared reflectance spectrometer(VECTOR22/N; BURKER Optik, Ettlingen, Germany), and spectra of the samples were converted into numerical values using the NIRS model of Xu et al.,the content of each of these traits was expressed in percent of dry matter[15].
The NIRS prediction equations for CEL,LIG,ADF,NDF,and IVDMD were built using the partial least squares method of OPUS 6.0 software [16]. The corresponding internal crossvalidation determination coefficients (R2CV) of the prediction model were 94.0%, 90.5%, 93.6%, 95.3%, and 90.2%. The external validation determination coefficients (R2Val) were 96.7%, 92.7%, 94.6%, 96.5%, and 91.2%. According to these parameters, the model can be used to predict stem cell-wallrelated traits well.
Because NDF is composed mainly of lignin, cellulose, and hemicellulose, which are constituents of the cell wall,cellulose and lignin were expressed as percentages of NDF(CEL/NDF and ADL/NDF,respectively).In vitro NDF digestibility(IVNDFD)was computed assuming that the non-NDF components of the cell wall were completely resolved [17,18]. The formula was IVNDFD = 100 × (IVDMD - (100 - NDF) / NDF).
2.3. Phenotypic data analysis
Phenotypic data processing was performed with Microsoft Excel 2016(https://products.office.com/zh-cn/home).Descriptive statistics for all six traits and correlations between traits were calculated with SPSS 20.0 (SPSS Inc., Chicago, IL, USA).Other analyses were implemented in R software,version 3.3.2(https://www.r-project.org/).
Analysis of variance was performed with the aov function in R. Lines, environments and replications were treated as random variables. The genetic variance (), genotype ×environment variance (), and random error () were used to calculate broad-sense heritability using the formula
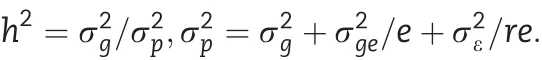
where, e = 2 environments and r = 3 replications in each environment. The 95% confidence intervals of the h2were computed[19].
The lmer function in the lme4 package of R was used to fit a mixed linear model to estimate the best linear unbiased prediction (BLUP)value for each trait for each line[20]:
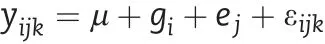
where, yijkis the kth observed value of the ith RIL in the jth environment, μ is the grand mean over all locations, giis the ith genetic effect,ejis the jth environmental effect,and εijkis a random error. In this model, μ was treated as a fixed effect and giand ejas random effects.
The BLUP values,representing the sum of fixed effects and estimated genotypic effects, were used for among-trait correlation analysis and QTL analysis.
The bivariate correlation analysis module in SPSS 20(SPSS Inc., Chicago, IL, USA) was used to analyze the correlation between phenotypes.The Pearson correlation coefficient was selected,and significance was tested with a paired t-test.
2.4. Development of genetic linkage map
Seedling leaves of the 188 RIL lines and parents were collected and ground into powder in liquid nitrogen.Genomic DNA was extracted using the CTAB method [21]. The DNA was genotyped with the MaizeSNP3K DNA-Chip [22], which contains 3072 SNPs, at the National Maize Improvement Center of China,China Agricultural University.
The genotype data were processed with PLINK [23] and JoinMap 4.0 [24]. PLINK was used for quality control of SNPs,and the following SNPs were deleted: SNPs with no polymorphism or heterozygosity between two parents,a missing rate≥20%, heterozygosity ≥20%, or minor allele frequency (MAF)≤0.05. A chi-square test was performed in JoinMap, and SNPs with partial separation were deleted.RILs with a missing rate≥20% or heterozygosity ≥10% were then deleted. Finally, a genetic linkage map comprising 756 SNPs was developed with QTL IciMapping 3.2 [25]. The steps were grouping (at a LOD threshold of 8.0), ordering, rippling, and outputting. The genetic distance between markers in centiMorgans was calculated with the Kosambi function.
2.5. QTL identification
QTL IciMapping 4.1 (http://www.isbreeding.net/software/?type=detail&id=18) was used for detecting the QTL location and effects of target traits,employing the inclusive composite interval mapping of additive QTL (ICIM-ADD) and intergenic epistatic QTL(ICIM-EPI)modules[25-27].The phenotypic data used were BLUP values, which have had environmental effects removed. For ICIM-ADD, the scanning step was set at 1.0 cM and the probability for entering variables(PIN)was set at 0.001 [26]. For ICIM-EPI, the scanning interval between markers was set at 5 cM, and the P-value for PIN was 0.0001[27]. In both modules, missing data were deleted, and LOD thresholds were determined by 1000 permutation tests at P <0.05 to test for type I error[28].The proportion of observed phenotypic variance explained by each QTL was estimated.The format for naming a QTL was “trait + chromosome +number”: for example, adf9-1, where adf represents the ADF trait, 9 represents the chromosome number, and 1 is the number of QTL on the ninth chromosome. Determining the confidence intervals for the locations of the QTLs by one-LOD support intervals to each side of the position of the maximum LOD.
3. Results
3.1. Phenotypic variation, correlations, and heritability
Phenotypic values of the six target traits for the parents and 188 RILs appear in Table 1. The mean values of By804 were higher than those of B73 for all traits but ADF and NDF, andonly the differences in ADL/NDF and CEL/NDF between the two parent lines were highly significant(P = 0.01;Table 1).The values of ADF and NDF in the RIL population were higher than those of the highest parent line, and the values for the other traits were between those of the parent.CV values were in the range 2.99%-16.39%, with variation ranging from 9.26%-21.00% for IVNDFD and 43.69%-52.13% for CEL/NDF based on the BLUP values. The six traits showed normal distributions(Fig.1).
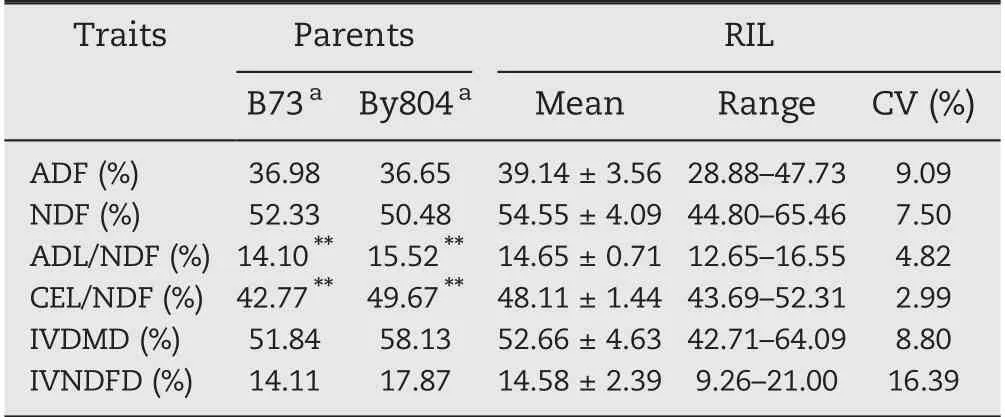
Table 1-Descriptive statistics for the six cell-wall-related traits of the maize stalk in the RIL population.
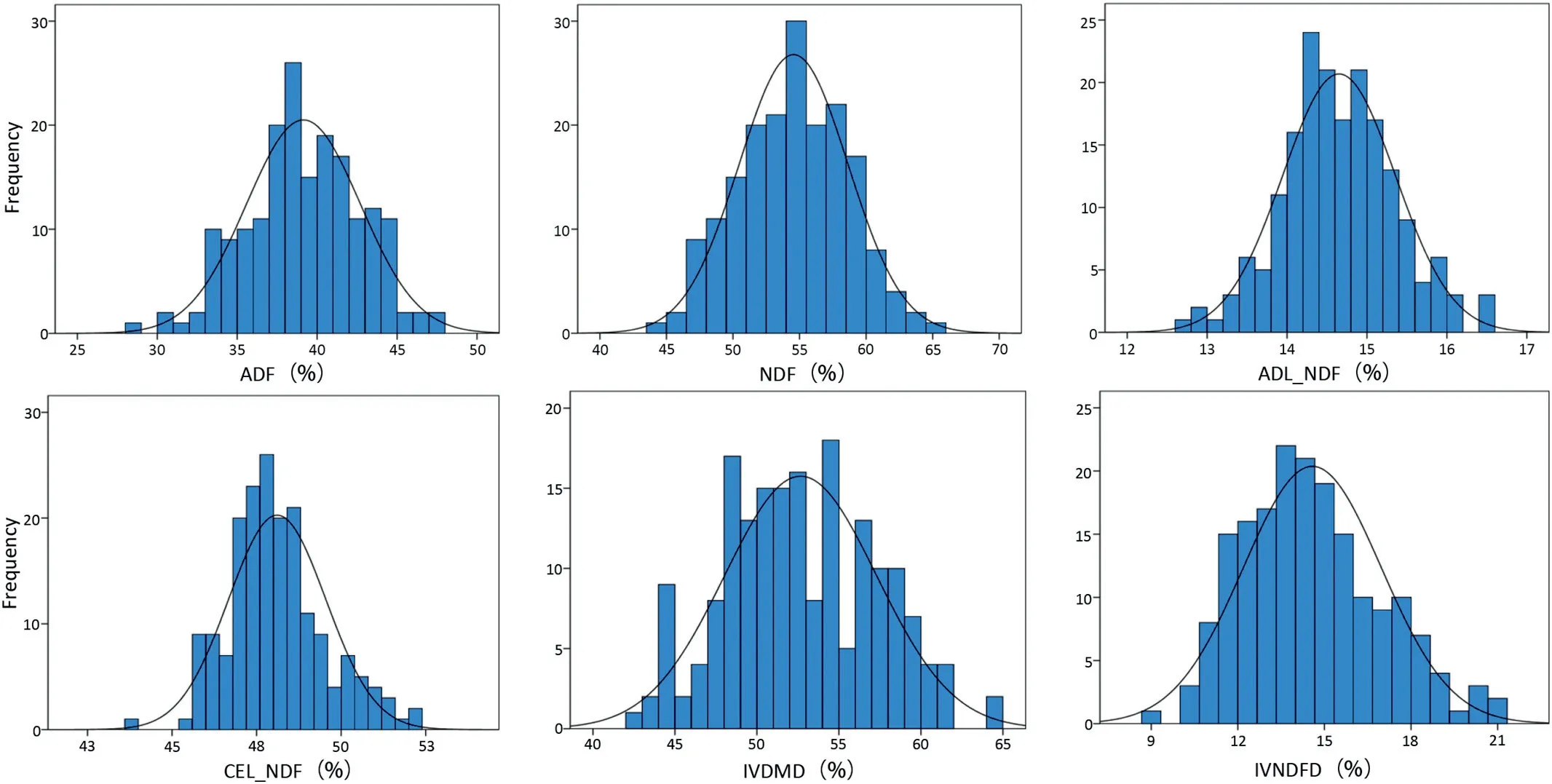
Fig.1- Distributions of the six cell-wall-related traits of the maize stalk in the RIL population.
The results of variance analysis show that the cell wall compositions and the digestibility of the maize stem were significantly affected by genotype, environment, and the interaction between genotype and environment, except for IVNDFD (Table 2). This result indicates that cell-wall-related traits are sensitive to genotype and environment. The broadsense heritability of the traits ranged from 0.52 (IVNDFD) to 0.73 (ADF), with the respective confidence intervals 0.38-0.62 and 0.65-0.79.
There were highly significant correlations among the six traits,with coefficients from 0.368 between IVNDFD and ADL/NDF to 0.971 between ADF and NDF (Table 3). ADF was significantly positively correlated with NDF and negatively correlated with the other traits. There were significant negative correlations between NDF and the other cell-wallrelated traits.Finally,ADL/NDF,CEL/NDF,IVDMD,and IVNDFD were significantly positively correlated with one another.
3.2. QTL mapping
After screening, 756 SNPs were used for constructing a genetic linkage map.These SNPs covered a total of 1607.7 cM,with a mean interval of 2.13 cM between markers(Table S1).
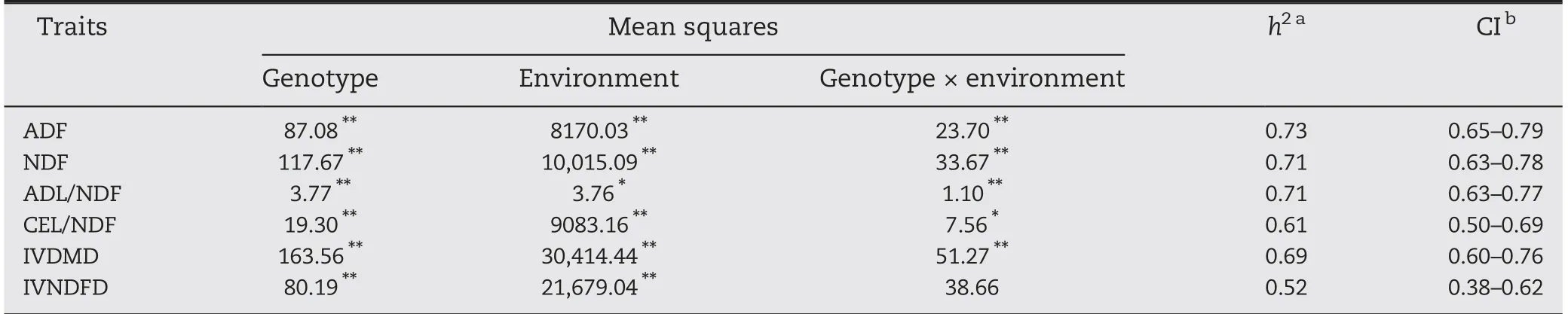
Table 2-ANOVA and broad-sense heritability for six cell-wall-related traits of the maize stalk in the RIL population.
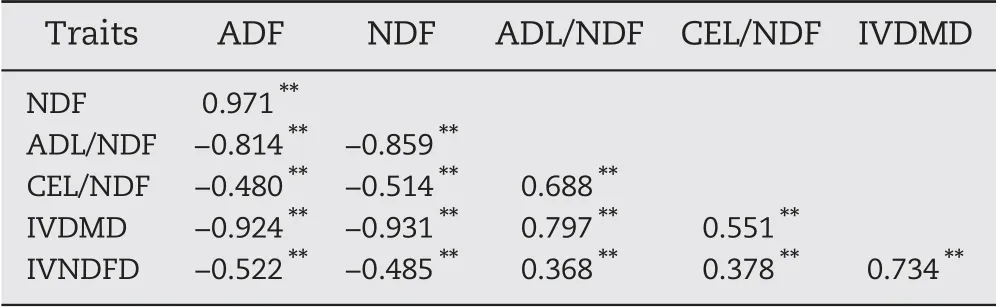
Table 3-Phenotypic correlations among six cell-wallrelated traits of maize stalk in the RIL population.
After 1000 permutation tests, the LOD threshold for ADF and IVDMD was determined to be 3.2,that for ADL/NDF,CEL/NDF, and IVNDFD 3.1, and that for NDF 3.0. A total of 20 QTL affecting cell wall components and digestibility traits were detected, of which 4 were for ADF, 5 for ADL/NDF, 3 for CEL/NDF, 2 for IVDMD, 1 for IVNDFD, and 5 for NDF. These QTL were located on chromosomes 1, 2, 6, 7, 8, and 10. The phenotypic variation explained by the QTL ranged from 1.52%to 20.01%, with significant differences. At least 1 major QTL accounted for >10% of the phenotypic variation for each trait(Table 4). The two major-effect QTL adf2 and adf6 explained 12.20% and 10.72% of the phenotypic variation, respectively.The additive effects of the B73 alleles at these two loci were 7.33%and 1.15%,respectively.
For ADL/NDF,the favorable alleles of three QTL(adl1,adl2-2, adl6) were from By804 and those of two (adl2-1, adl7)from B73. The QTL of adl2-1, mapped on chromosome 2,accounted for the largest effect, explaining 20.01% of phenotypic variation. Another primary QTL, adl2-2, adjacent to adl2-1, explained 10.30% of phenotypic variation. The remaining three minor effect QTL explained <7% of phenotypic variation.
Three QTL controlling CEL/NDF were distributed on chromosomes 1 and 6 and explained a total of 29.31% of phenotypic variation. The two major QTL cel1 and cel6-2 explained respectively 12.43% and 10.48% of phenotypic variation. The alleles of these two QTL associated with an increase in ADL/NDF originated from By804.
Only 2 and 1 QTL, located on chromosomes 2, 6, and 10,significantly affected IVDMD and IVDFD, accounting for respectively 22.37% and 10.01% of phenotypic variation. The alleles of ivdmd6 and ivndfd10 with additive effects came from By804, and the allele of ivdmd2 from B73 contributed to increase IVDMD.
Five QTL were detected for NDF on chromosomes 2, 6, 7,and 8 and explained a total of 41.14% of the phenotypic variation. Among these QTL, the ndf2-1 allele from By804 accounted for the highest explained phenotypic variation(13.36%). The other QTL explained 6.03%-7.95%of phenotypic variation.B73 alleles at ndf6 and ndf8 loci and By804 alleles at ndf2-2 and ndf7 loci increased NDF concentration.
No epistatic QTL were detected.
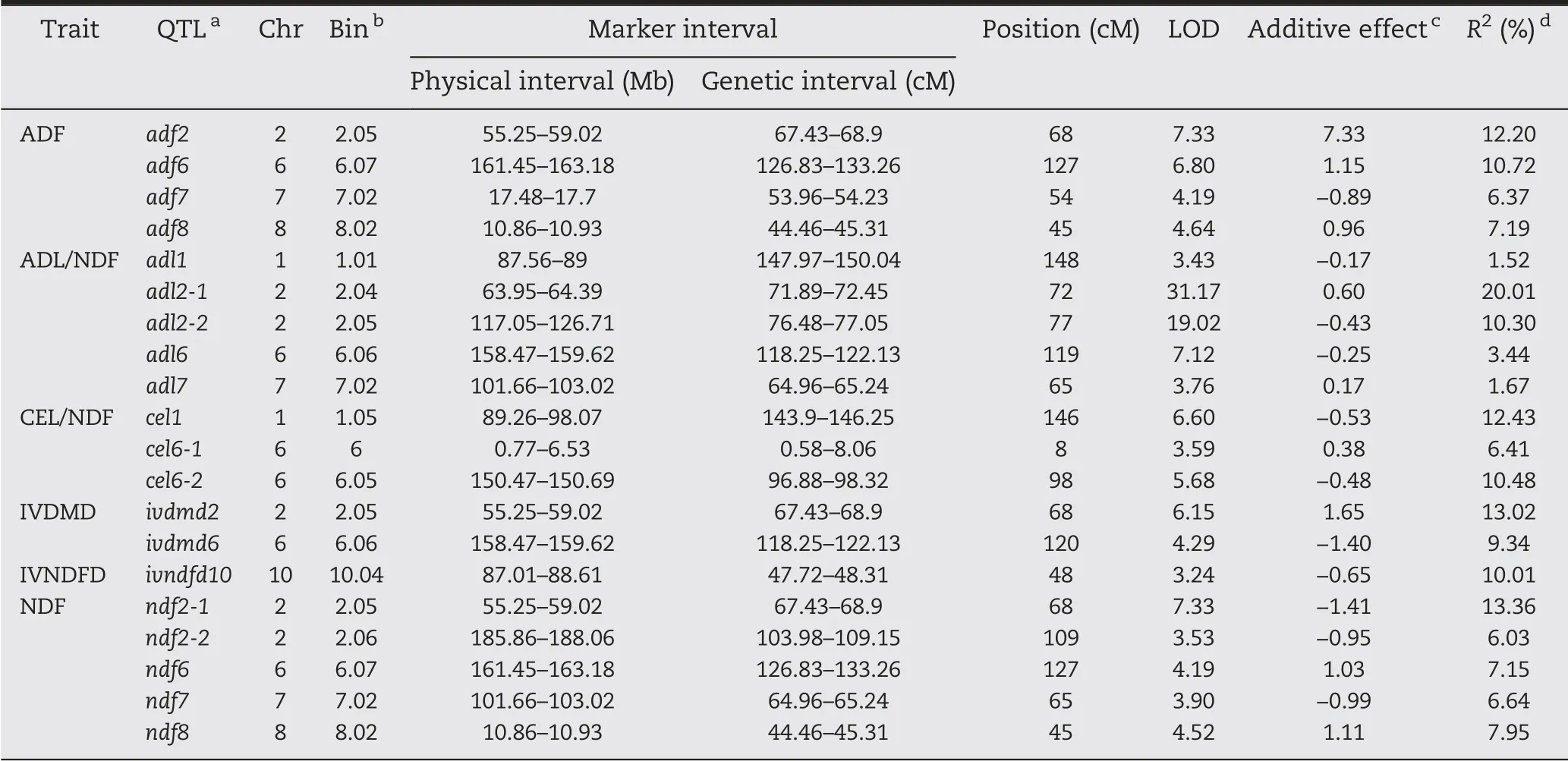
Table 4-QTL for six cell-wall-related traits of the maize stalk in the RIL population.
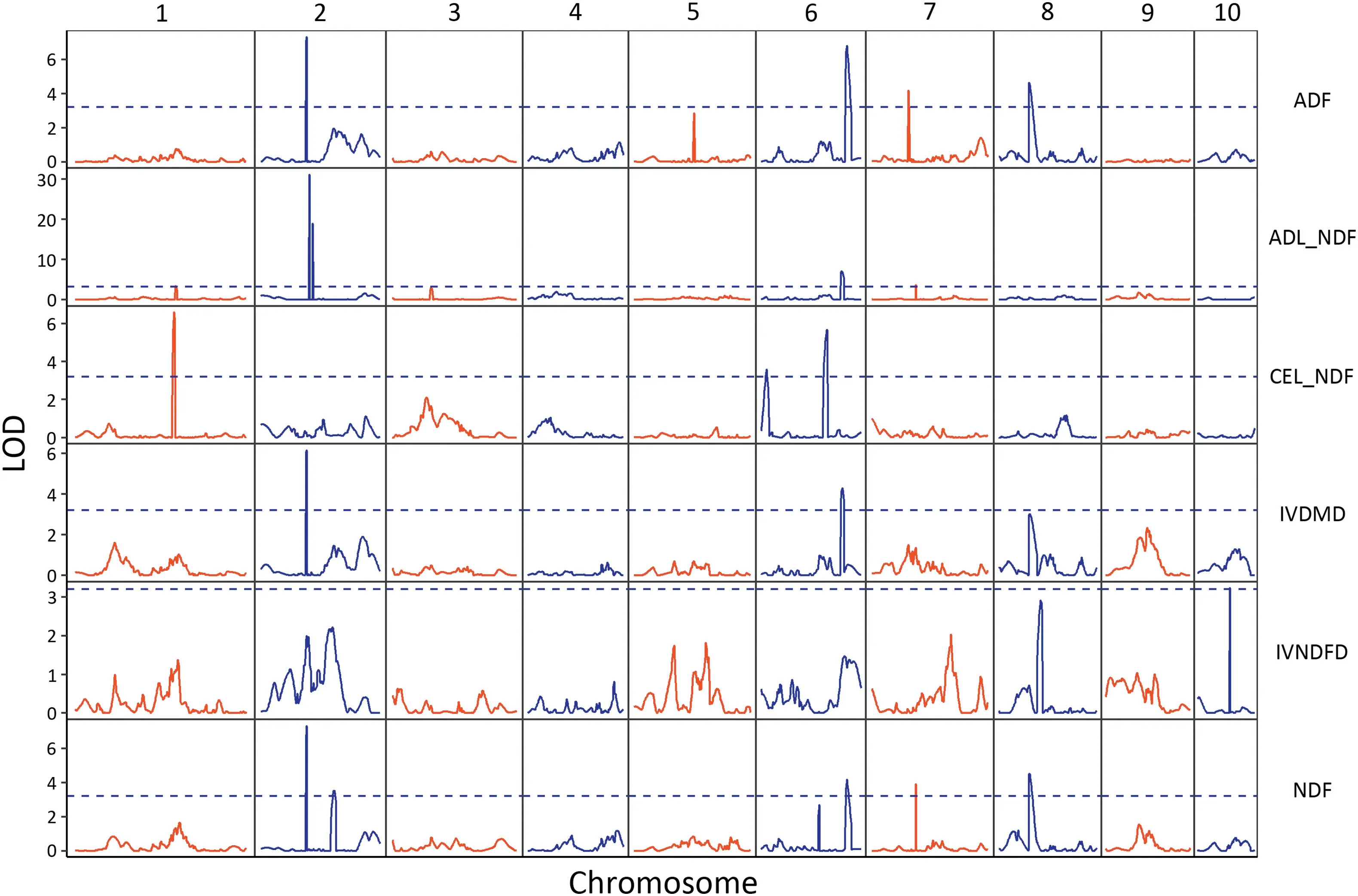
Fig. 2 - Distribution of detected QTL for six cell-wall-related traits of the maize stalk in the RIL population.
3.3. Clusters of QTL
A few QTL controlling different traits were located in the same chromosome regions(Fig.2).On chromosome 2,QTL for ADF,IVDMD and NDF shared the same genetic interval,from 55.25 to 59.02 Mb. All of them were large-effect QTL. The QTL adl6,ivdmd6 and adf6,and ndf6 were located in adjacent confidence intervals. One region spanning 101.66-103.02 Mb on chromosome 7,contained adl7 and ndf7.The additive QTL for ADF and NDF(adf8 and ndf8)were mapped in the same genetic interval of chromosome 8,namely 10.86-10.93 Mb.
4. Discussion
4.1.Phenotypic variation,heritability,and correlations of traits
Except for ADF and NDF content, the mean cell wall digestibility of the high-oil maize inbred line By804 was higher than that of the common line B73, as was reported by Wang [7] using the high-oil maize inbred line Ce03005 (lower fiber content and higher digestibility) and B73 (higher fiber content and lower digestibility). It could be conjectured that high-oil maize possesses higher forage quality than common maize. According to the BLUP values, the cell wall compositions and digestibility traits in this investigation were different from previous results [3,29-36]. For example, the ADL/NDF ranging from 12.65% to 16.55% in this study was higher and the IVNDFD ranging from 9.26% to 21.00% was lower than in previous reports.These differences may be due to variations in test materials and population size.
The finding that all traits followed normal distributions and showed transgressive segregation suggests that they are quantitative traits controlled by multiple genes. Cell wall components and digestibility were significantly affected by genetic and environmental factors, and the broad-sense heritabilities of each trait were moderate (IVNDFD: 0.52) to high (ADF: 0.73). There were significant correlations between each pair of cell-wall-related traits. Previous reports showed the same correlations between ADF, NDF, IVDMD, and IVNDFD [31,37]. However, Mechin et al. [29] found that NDF was negatively correlated,but not significantly so,with IVDFD in a population consisting of hundreds of RILs developed from a cross between Io and F2. Other studies [35,38] have shown significant negative correlations between CEL/NDF and IVNDFD as well as between ADL/NDF and IVNDFD,in contrast to our finding of positive correlations between these traits.These differences demonstrate the complex relationship between cell wall compositions and digestibility. Digestibility is affected by content and composition of the cell wall components. The accumulation of cell wall components is a dynamic process and the correlations between the traits differs at different developmental stages. Our results apply only to the silking stage. How the correlation between cellwall-related traits would change with the progression of development awaits further study.
4.2. QTL analysis
Among the QTL detected in this study,only one was observed to control IVNDFD, possibly because the large environmental effect on IVDFD led to a relatively low heritability. The QTL ivndfd10 was also mapped to the same genetic position (bin 10.04)in a population of 240 RIL derived from F838 × F286 and accounted for 9.6%of phenotypic variation[32].The QTL adl2-1(explaining 20.01%of the phenotypic variation for ADL/NDF),was also mapped to the same genetic position(bin 2.04)in two previous studies[33,36],where it explained respectively 17.7%and 19.5% of phenotypic variation. Further QTL, namely adl1,adl7,ndf2-1,ndf7,and ndf8,have also been reported previously[3,31,36,37,39]. QTL with large or stable effects can be considered priority candidate sites for fine mapping using near-isogenic lines(NIL)and can then be expected to be used in marker-assisted breeding.
Most of the QTL mapped in this study have not been previously reported.No epistatic QTL were detected,whereas in a previous study [12], cell wall-related traits were affected by relatively small epistatic effects in addition to major and dominant effects. These differences may be due to the use of different parent materials,selected tissues,and growth stages in addition to population size and molecular marker type.The high-oil maize inbred line By804 material used in this study was not used in previous studies[37].
4.3. Co-localization of QTL for cell wall components and digestibility
Many QTL associated with cell wall components and digestibility of the maize stalk were located in common genome regions,including bins 2.05, 6.06, 6.07, 7.02, and 8.02. The QTL for ADF and NDF were both mapped to bins 6.07 and 8.02 and the alleles of these loci showed positive effects from B73.The QTL for ADL/NDF and IVDMD were both located in bin 6.06,and the positive alleles of these two loci were both from By804. These results suggest that some genes controlling cell wall compositions and digestibility may be the same(pleiotropic)or genetically linked.Perhaps these genes are responsible for the correlations between the cell-wall-related traits.It was shown in a previous study [40] that phenotypic correlations between quantitative traits may derive from the correlation between QTL controlling them. The alleles of the loci co-localized in bins 2.05 and 7.02 showed positive effects from different parents.In other studies,some QTL hotspots were the same and some were different[41-43]. For example, Ralph et al. [41] and Barrière et al. [42]identified one common hotpot (bin 6.06). It was shown [37,43]that some QTL in the hotspot regions were located in the same chromosomal regions of candidate genes or related QTL that directly or indirectly control cellulose or lignin biosynthesis(e.g.,ZmCesA7, ZmCesA9, and CCR2 in bin 7.02). The regulation of these QTL may also account for the correlations between cellwall-related traits.
Further studies are needed to identify overlapped regions more accurately. It will then be necessary to optimize the marker density and improve the linkage map, laying the foundation for the fine mapping of QTL and providing theoretical basis for increasing forage quality by marker-assisted selection.
Supplementary data for this article can be found online at https://doi.org/10.1016/j.cj.2019.06.009.
Declaration of competing interest
The authors declare that they have no competing interests.
Acknowledgments
This study was supported by the National Key Research and Development Program of China (2017YFD0101201 and 2016YFD0101002), the Chinese Academy of Agricultural Sciences through the Agricultural Science and Technology Innovation Program (CAAS-ASTIP-2017-TRICAAS) and National Engineering Laboratory for Crop Molecular Breeding.

- The Crop Journal的其它文章
- Brief Guide for Authors
- Rapid generation advance(RGA)in chickpea to produce up to seven generations per year and enable speed breeding
- Transcriptomic responses in resistant and susceptible maize infected with Fusarium graminearum
- Relay-intercropping soybean with maize maintains soil fertility and increases nitrogen recovery efficiency by reducing nitrogen input
- Genetic bases of source-, sink-, and yield-related traits revealed by genome-wide association study in Xian rice
- Performance and yield stability of maize hybrids in stress-prone environments in eastern Africa