
Extensive exploration of T cell heterogeneity in cancers by single cell sequencing
2019-05-25 07:49:54XiaofangWangYangqiuLi
Xiaofang Wang, Yangqiu Li
1Department of Hematology, First Affiliated Hospital, School of Medicine, Jinan University, Guangzhou 510632, China;
2Key Laboratory for Regenerative Medicine of Ministry of Education, Institute of Hematology, School of Medicine, Jinan University, Guangzhou 510632, China
Abstract Human T cells are a highly heterogeneous population and can recognize a wide variety of antigens by their T cell receptors (TCRs). Tumor cells display a large repertoire of antigens that serve as potential targets for recognition,thus making T cells in the tumor micro-environment more complicated. Making a connection between TCRs and the transcriptional information of individual T cells will be interesting for investigating clonal expansion within T cell populations under pathologic conditions. Advances in single cell RNA-sequencing (scRNA-seq) have allowed for comprehensive analysis of T cells. In this review, we briefly describe the research progress on tumor microenvironment T cells using single cell RNA sequencing, and then discuss how scRNA-seq can be used to resolve immune system heterogeneity in health and disease. Finally, we point out future directions in this field and potential for immunotherapy.
Keywords: T cells; tumor micro-environment; single-cell RNA-sequencing
Introduction
Human immune system is composed of multiple cells that coordinately protect against various pathogens. Among these cells, T cells recognize a broad range of self and foreign antigens via a large heterogeneous population of surface antigen receptors, the T cell receptors (TCRs).TCRs vary from cell to cell and represent a sort of“molecular tag” for T cells. T cells are key elements of cancer immunology and T cells found in the tumor microenvironment are named tumor-infiltrating lymphocytes (TILs) (1). In a subset of solid tumors(colorectal cancer, breast cancer, renal cell carcinoma,ovarian cancer, and gastrointestinal stromal cancer),activated CD8+T cells in the tumor microenvironment have positive prognostic value (1-8).
However, increasing data show that T cell tolerance is associated with a failure in antitumor immunity. Firstly,Treg cells prevent cytotoxic T cells activity and promote tumor growth by secreting a volume of T cell suppressive molecules, including IL-10, IL-35, and TGF-β (9,10).Furthermore, TIL cells are regulated by a complex immunosuppressive network, which drives T cells to terminally exhausted T cells (11). T cell exhaustion increase high levels of inhibitory receptors such as programmed cell death protein 1 (PD-1), lymphocyte activation gene 3 protein (LAG-3), cytotoxic T lymphocyte antigen-4 (CTLA-4), and T-cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain(TIGHT) (12-17). Impaired production of effectors, such as IL-2, TNF-α, TFN-γ and GzmB, also are demonstrated by exhausted T cells (18). Thus, therapeutic strategies to overcome T cell exhaustion and modulate Tregs are key for solving immune escape by cancers.
The success of cancer immunotherapies based on CTLA-4 or PD-1 blockade has been attributed to the prevention of T cell exhaustion (19). These treatments have been successful in treating melanoma, lung cancer,and kidney cancer (20,21). However, effective responses have only been observed in a subset of patients and cancer types. The variation in treatment efficacy is linked to the heterogeneity in the immune cell composition of individual tumors in individual patients. It has been reported that significant heterogeneity in the immune composition is observed across tumor subtypes and patients with breast cancer (22). Thus, the question becomes whether T cell state could be an indicator of treatment and prognostic markers. To better define T cell subtypes in tumors, largescale and high-dimensional analyses are needed.
Application of single cell RNA-sequencing(scRNA-seq) to study heterogeneous T cells
Flow cytometry, immunohistochemistry and bulk RNA analysis using a limited number of established markers cannot meet the needs of solving issues surrounding heterogeneity in the T cell composition of individual tumors. scRNA-seq is a powerful technology for resolving heterogeneous cell types and states in human tissues such as the intestine (23,24), brain (25), spleen (26), pancreas(27,28), and blood (29-31). scRNA-seq provides insights into cell functional heterogeneity that were hidden in the bulk cell population. Nowadays, the application of this technique to immune profiling has begun to be realized(32-34). In different tumor micro-environments, different T cell subtypes can be influenced and display several dominant TCR display clones. In addition to transcriptome analysis, the development of scRNA-seq technologies has opened new perspectives for TCR analysis. Studying paired TCR sequences in individual cells will be powerful for understanding the adaptive immune response. Individual phenotypic states are likely shaped by antigenic TCR stimulation and environmental stimuli. Furthermore,TCRs can be used to monitor the dynamics of T cells in tumor immunology (32,35). Thus, making a connection between TCR clonotype composition and the transcriptional landscape of individual T cells will be interesting for investigating the dynamics of clonal expansion within T cell populations under normal or pathologic conditions.
Fluorescence-activated cell sorting (FACS)-purified CD45+CD3+cell populations are commonly subjected to sequencing. 10X Genomics or SAMRT-seq2 is used to isolate cells and amplify transcripts (36-39). 10X Genomics captures cells using microfluidic devices that trap cells inside a hydrogel. SMART-seq2 relies on FACS into 96-well plates that physically separate one cell from another.Once cells are lysed, sequencing protocols are characterized by a reverse transcription and amplification step before library preparation. The scRNA-seq data generated using 3’/5’ or full-length approaches have been exploited to extract information about T cell properties and heterogeneity. The assembly of full-length (TCR)sequences from scRNA-seq data allows for alpha and beta chain sequence pairing, which is not possible when performing “bulk” analyses, and for the integration of clonality information using the transcriptome of a single T cell (Figure 1). In Table 1, we summarize the methods of scRNA-seq that are used or potential to be used in T cell analysis.
scRNA-seq of T cells in tumors
Many research groups have studied transcriptional maps of T cells in different tumors using scRNA-seq. Here, we summarize several pioneering studies that highlight how scRNA-seq can be used for the discovery of T cell cellular states in tumors.
Breast cancer
Breast cancer is the most common cancer in women.Although breast cancer has not been considered as a cancer for the application of immunotherapeutic treatments,recent studies have demonstrated evidence that the immune system plays a complex role in breast cancer biology by promoting tumor growth and mediating the eradication of this disease. TILs are significantly increased in a subset of patients, including triple-negative and HER2-positive breast cancers (22,42), making them a prognostic marker for chemotherapy and survival.
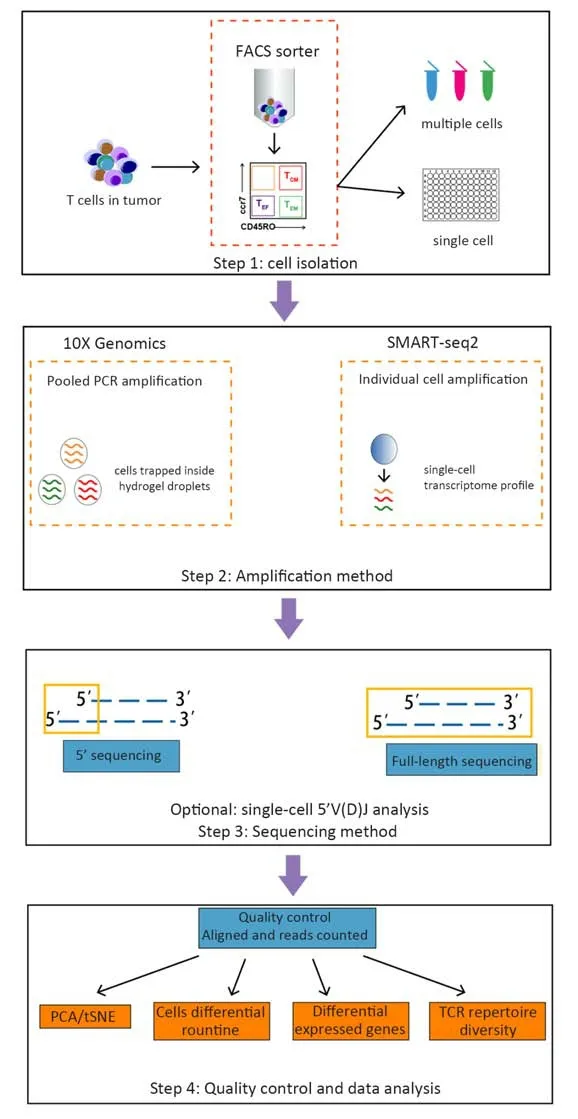
Figure 1 Overview of scRNA-seq technology in study of T cells. T cells are usually isolated from tumors using fluorescence-activated cell sorting (FACS). Step 1: Cell isolation. 10X Genomics: first isolate T cells and then capture cells using microfluidic devices that trap cells inside hydrogel droplets; SMART-seq2: isolate individual cells into 96-well plates and profile single-cell transcriptomes separately. Step 2:Amplification method. In this step, T cells are lysed, and reverse transcription and PCR amplification are performed. 10X Genomics uses cellular barcoding techniques and only one PCR reaction, while SMART-seq2 requires the number of PCR reactions to equal to the number of profiled cells. Step 3: Sequencing method. 10X Genomics uses sequencing of the 3’/5’ end of each transcript while SMART-seq2 can sequence full-length transcripts. Either can choose to analyze single-cell 5’ V(D)J regions. Step 4: Quality control and data analysis.
Recently, researchers from Memorial Sloan Kettering Cancer Center profiled 45,000 immune cells from 8 breast carcinomas and matched normal breast tissue, blood, and lymph nodes. This group derived an immune map of breast cancer, pointing to continuous T cell activation and differentiation states (35). In agreement with previous reports (22,43), these researchers found an activation gradient in CD8+T cells in tumors. Moreover, T cells in the blood and lymph nodes exhibited varying phenotypes compared with T cells in breast tissue. Tumor-resident T cells, including CD4/CD8 effector cells, central memory clusters and Treg clusters, might be exposed to varying degrees of inflammation, hypoxia and nutrient deprivation,thus exhibiting the expression of anti-inflammatory,exhaustion, hypoxia and anergy genes. Further analysis of paired single-cell RNA and TCR sequencing data from 27,000 additional T cells revealed the combinatorial impact of the TCR on phenotypic diversity. When analyzed in conjunction with TCR utilization, it appears that T cell populations are associated with unique combinations of TCR clonotypes. In addition, these TCR patterns together with unique gene expression programs and environmental exposure define the states of intratumoral T cells.

Table 1 Summary of current scRNA-seq methods used or potential to be used in T cells analysis
Another group from Melbourne later performed the same profiling of tissue-resident memory T cells (TRM) in breast cancer (44). Although only examining two tumors,they identified five CD4+and four CD8+T cell clusters,including an unexpected CD8+TRM-like population. CD8+TRMcells have been reported to be very effective cytokine producers and superior to TEMcells in their re-infection response (45). TRMcells in human breast cancer tissue expressing more granzyme B transcripts than the TEM-like population. Furthermore, a subset of the TRMpopulation appeared to be proliferating based on the expression of cell cycle genes, indicative that the population is expanding in response to tumor antigens. The authors further substantiated this finding by bulk RNA sequencing of FACS sorted CD103+and CD103-populations of CD8+T cells, finding significantly higher granzyme B and perforin expression in the CD103+population, implying a greater cytotoxic function. Thus, CD8+TRMcells contribute to breast cancer immune surveillance and may be key targets for modulating immune checkpoint inhibition.
Lung cancer
Non-small-cell lung cancer (NSCLC) accounts for approximately 85% of lung cancer cases and is the leading cause of cancer-related mortality (46). Continuing clinical responses can be obtained in NSCLC when using immunotherapies (47-49); however, efficacies vary partially due to the amounts and properties of TILs (50-52). To examine this problem, Zemin Zhang and colleagues dissected the landscape of TILs from 14 untreated NSCLC patients (53). The researchers observed two clusters of cells exhibiting states preceding exhaustion and a high ratio of“pre-exhausted” to exhausted T cells was associated with better prognosis. Furthermore, Tregs with TNSFSR9 and IL1R2 correlate with poor prognosis. By examining genes that are cancer immunotherapy targets in clinical trials, it was found that genes are in the category of effector reactivation or anti-Treg. By dissecting the TCR repertoire, it was concluded that Treg cells do not clonally enrich in tumors, suggesting recruitment from the periphery, and CD8+T cells are clonally enriched, which points to clonal activation and expansion inside tumors.
At the same time, Berbard Thienpont and colleagues reported the tumor microenvironment (TME)transcriptome in human NSCLC (54). This analysis included immune cells (myeloid, T and B cells), fibroblasts,endothelial cells, alveolar cells, and epithelial cells but not cancer cells. This group found that CD8+and regulatory T cells are enriched in tumors, while CD4+and natural killer cells appear to be depleted. Higher cytotoxic activities are curtailed by high checkpoint expression in CD8+T cells.Cell proliferation pathways were generally lower in TILs and CD8+T cells; however, there was accordingly increased oxidative phosphorylation and fatty acid oxidation (55). In this case, targeting these pathways may enhance immunotherapy and provide exciting avenues for the design of novel therapies.
Liver cancer
Hepatocellular carcinoma (HCC) is the most prevalent tumor type in the liver, and it is often related to chronic hepatitis B virus infection, obesity, or alcohol. However,there are limited treatment options and a lack of clinical success in immunotherapies (56). TILs in HCC are significantly increased, but they are dysfunctional (57). To characterize the TILs in HCC, Zemin Zhang’s group analyzed greater than 5,000 single T cells isolated from HCC patients and studied their complete TCR sequences and transcriptomes simultaneously (58). A total of 11 unique T cell subtypes, including 5 CD8+and 6 CD4+T cell sub-clusters could be distinguished by gene expression.This group found that exhausted CD8+T cells and Tregs are enriched and clonally expanded in HCC. Primary CD8+T cells, which overexpress layilin (LAYN), resulted in interferon (IFN)-γ reduction, CD4+Treg cell suppression, and CD8+T cell exhaustion as a response.IFN-γ is required for active tumor cell killing, and LAYN is associated with exhausted CD8+TILs and poor prognosis. Based on the clonal TCR data, it appears that most of the Treg cells in HCC are unique clones, and CD8+T cells more likely evolve from other types of CD8+T cells inside HCC tumors. Targeting the layilin pathway may be a potential avenue for HCC treatment in the future.
Colorectal cancer
Recent clinical trials have demonstrated that patients with colorectal cancer who display microsatellite instability(MSI) but not microsatellite-stable (MSS) phenotypes respond to the immune-checkpoint blockade of PD-1 (59).Zhang’s group used STARTRAC, an integrated approach of RNA sequencing and TCR tracking, to obtain transcriptomes of 11,138 single T cells from 12 patients with colorectal cancer. They quantitatively analyzed the dynamic relationships among 20 identified T cell subsets with distinct functions and clonalities (60). It showed that both CD8+effector and “exhausted” T cells exhibited high clonal expansion. Most CD4+TIL cells and Treg cells showed clonal exclusivity, whereas certain Treg cell clones were linked to several T helper cell clones. It’s noticed that two IFNG+Th1-like cell clusters were identified with distinct IFN-γ-regulating transcription factors. One is the GZMK+effector memory T cells, which were associated with EOMES and RUNX3. The other is CXCL13+BHLHE40+Th1-like cell clusters, which were associated with BHLHE40. Only CXCL13+BHLHE40+Th1-like cells were enriched in colorectal cancer patients with MSI, and this could explain their responses to immune-checkpoint blockade. Furthermore, IGFLR1 was highly expressed in both CXCL13+BHLHE40+Th1-like cells and CD8+exhausted T cells and possessed costimulatory functions. These distinct populations can be used as a resource for T cell exploration and therapeutic targets.
The findings of TILs are summarized in Table 2.
Conclusions
Before the development of scRNA-seq, the discovery of new cell subsets involved using cell surface markers.scRNA-seq opens up a new era in which we can now construct a T cell atlas from normal and tumor tissues or cancerous lymph nodes, thus revealing the diversity in the tumor micro-environment, discovering variations in gene expression, and building developmental trajectories. In sum, T cells are heterogeneous in tumors, and Tregs are usually enriched, while CD8+T cells become exhausted TILs or TRMcells in tumors. Anti-inflammatory,exhaustion, hypoxia and anergy genes are up-regulated in these cells, and cancer immunotherapy target genes exhibit distinct expression patterns among the cells. In this manner, different T cell populations could be targeted by different immunotherapies. The T cell composition may be an important biomarker for subtyping tumors and an indicator for patient treatment.
It is important to note that the above-mentioned T cells are αβ T cells, but there is another T cell subtype that occupies 5%-10% of all peripheral T cells in healthy individuals called γδ T cells (61). Unlike αβ T cells, γδ T cells target cancer cells without requiring MHC presentation. γδ T cells have been studied for their therapeutic potential in cancer treatment (62). In this way,taking advantage of scRNA-seq in underlying γδ T cell immune states in tumors is vital for developing strategies for immunotherapy.
Although mouse models have been shown to be useful for understanding immune system effects, scRNA-seq allows for the profiling of human tissues. Furthermore, the diversity of T cells is not only reflected in transcriptome differences, activation state ranges, and TCR clone type diversity, it is also reflected in methylation and chromatin.We anticipate that the development of new technologies will extend single-cell profiling beyond the transcriptome with advances in methylation (63-66), proteome (67),chromatin (68,69), and genome (70) assays. Single-cell multi-omics analysis of individual cells will take place in the near future and help better understand the biology of immune-mediated diseases and monitor immune responses in cancer and post-therapies.
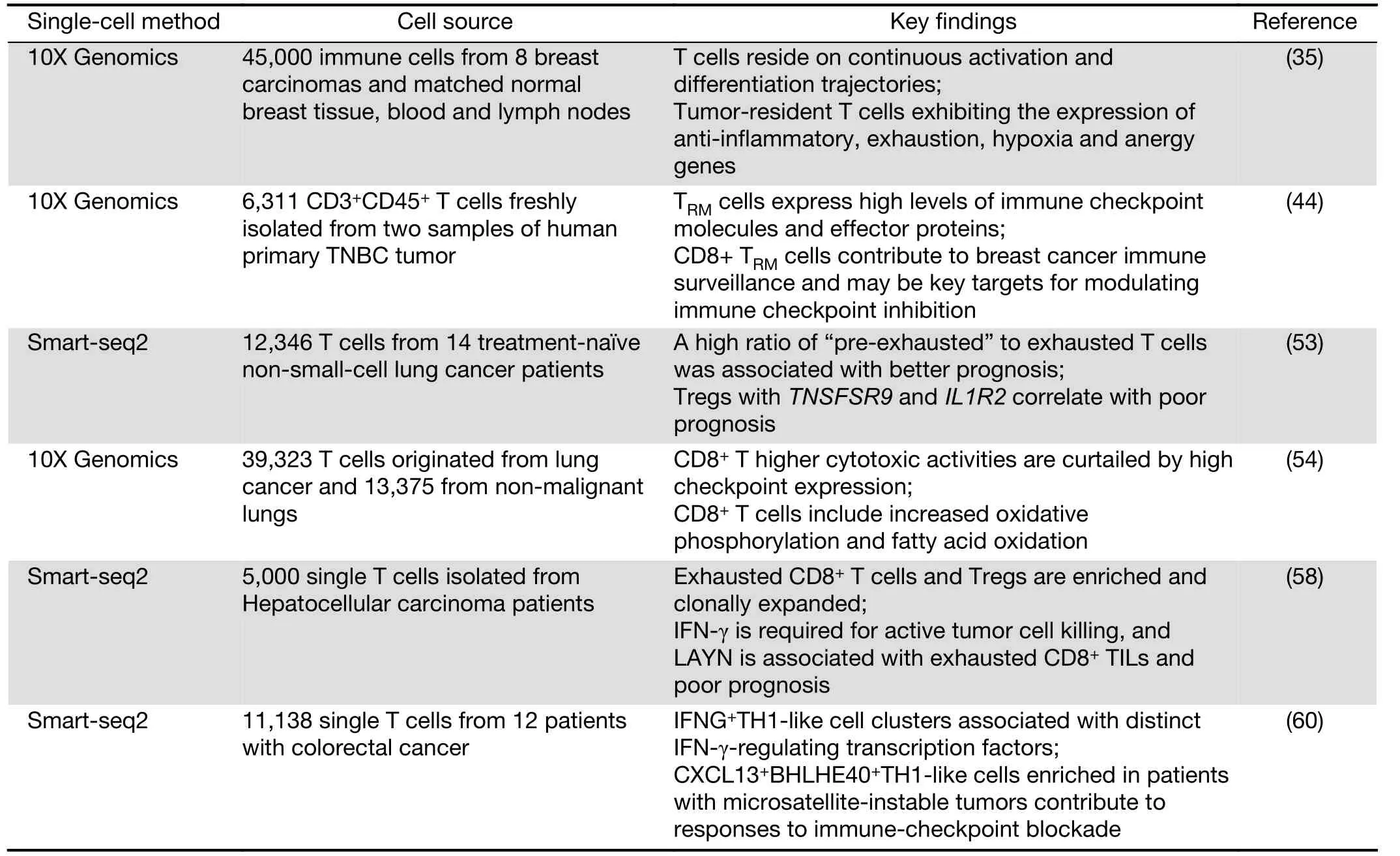
Table 2 Summary of recent study of T cells in tumors using scRNA-seq
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 81770152,91642111 and 81570143) and the Guangzhou Science and Technology Project (No. 201510010211, 201807010004 and 201803040017).
Footnote
Conflicts of Interest: The authors have no conflicts of interests to declare.
 Chinese Journal of Cancer Research2019年2期
Chinese Journal of Cancer Research2019年2期
- Chinese Journal of Cancer Research的其它文章
- Synthesis and evaluation of 64Cu-radiolabeled NOTA-cetuximab(64Cu-NOTA-C225) for immuno-PET imaging of EGFR expression
- Nomograms based on HPV load for predicting survival in cervical squamous cell carcinoma: An observational study with a longterm follow-up
- Alisol B 23-acetate-induced HepG2 hepatoma cell death through mTOR signaling-initiated G1 cell cycle arrest and apoptosis: A quantitative proteomic study
- Histogram analysis of apparent diffusion coefficient predicts response to radiofrequency ablation in hepatocellular carcinoma
- Efficacy of endoscopic treatment on patients with severe dysplasia/carcinoma in situ of esophageal squamous cell carcinoma: A prospective cohort study
- Sequential therapy according to distinct disease progression patterns in advanced ALK-positive non-small-cell lung cancer after crizotinib treatment