
A case of probable sporadic Creutzfeldt–Jakob disease
2018-01-08 09:25:11NoritaHussein
Norita Hussein
Department of Primary Care Medicine, Faculty of Medicine,50603, Kuala Lumpur, Malaysia E-mail: norita@ummc.edu.my
A case of probable sporadic Creutzfeldt–Jakob disease
Norita Hussein
Department of Primary Care Medicine, Faculty of Medicine,50603, Kuala Lumpur, Malaysia E-mail: norita@ummc.edu.my
Creutzfeldt-Jakob Disease (CJD) is a fatal neurodegenerative brain disease. The author describes a case presented to primary care clinic whereby neuropsychiatric symptoms were the patient’s initial presentation which later manifested with declining cognitive impairment, myoclonus and extrapyramidal symptoms. A typical abnormal magnetic resonance imaging (MRI) features were observed. The patient succumbed within six months of presentation.
Creutzfeldt-Jakob disease; neurodegenerative brain disease; neuropsychiatric
Background
Creutzfeldt—Jakob disease (CJD) is a rare human prion disease that is progressive and invariably fatal. The Centers for Disease Control and Prevention (CDC) Surveillance Unit reported one case per million population will develop annually [1]. The causative agent is the prion protein, and this disease has a long incubation period. Typically, sporadic CJD is characterized by rapidly progressive cognitive impairment often with myoclonus and cerebellar, extrapyramidal, or neuropsychiatric symptoms [1]. There is limited literature on CJD in Asia, perhaps being underreported or underdiagnosed. Case studies and case series have been reported in Malaysia [2], Singapore [3], Thailand [4],Taiwan [5], and Korea [6].
This case study describes probable sporadic CJD encountered in primary care. It is a challenge to diagnose at presentation because CJD is a rare disease and hence there was low index of clinical suspicion at the first encounter.
Case presentation
A 53-year-old woman presented to our clinic with difficulty to speak. Six months earlier, her family members had noted a progressive change in her personality and behavior; she was described as having become quieter than usual and withdrawn.Her interest in social activities had diminished and she had slowly withdrawn herself from her family members and neighbors.She used to be a socially active and a friendly person. Her husband noticed that she was not doing her routine housework and cooking anymore. She was seen crying inappropriately at times when alone in her room. Later, she developed progressive memory loss; for example, she forgot her children’s names and sometimes could not recall what she had eaten for meals that day. During this time, she was still able to talk and was ambulating well in the house.A month before the clinic visit, her gait had become unsteady and she had difficulty in coordination, with abnormal jerky limb movements noted at times. She then developed difficulty to speak, which made her seek medical treatment.
There was no history of fever, seizure, trauma, or any recent travel. There was no significant family history of a similar condition. She had diabetes and hypertension, which was well controlled, and her general health had been good.
Physical examination revealed a small-built woman who looked depressed. She had poor eye contact with minimal facial expression, had blank stares at times, and would cry inappropriately during consultation. She was disorientated to time, place, and person, and had difficulty to initiate speech.Her speech was soft and slurred. There were hand tremors,and brief myoclonic jerks were observed predominantly on her face and upper limbs during the consultation. There was muscle cogwheel rigidity on the upper and lower limbs. The reflexes and plantar responses were normal. She was not able to walk, and had truncal ataxia with unsteadiness, dysdiadochokinesia, and abnormal past-pointing. Other examination findings were unremarkable.
Her baseline investigations of full blood count, renal,liver, and blood chemistry, autoimmune screen, and blood culture revealed no abnormalities. Her chest radiograph and initial computed tomography scan of the brain were unremarkable.
The case was referred to a neuromedical specialist, and she was admitted for further treatment. Magnetic resonance imaging (MRI) including T2-weighed imaging and diffusionweighted imaging (DWI) was done immediately. The findings were consistent with CJD as shown in Fig. 1. Unfortunately,cerebrospinal fluid analysis was not done as family members refused lumbar puncture. The patient’s condition deteriorated while she was in the ward. She developed akinetic mutism within 1 week of admission; she was not able to speak and move at all and needed assisted ventilation. She remained akinetic and died after one month of hospitalization.
Discussion
There are three major types of CJD. It can occur in sporadic,familial, and acquired forms. The commonest form is sporadic CJD, which accounts for about 85% of cases [7]. The diagnosis of CJD is difficult. This case illustrates an uncommon early manifestation of sporadic CJD. This patient experienced personality and behavioral changes in the early course of the disease. These may be mistaken as an unusual psychiatric illness. The commoner neurological presentations of sporadic CJD occurred only at a later stage in this patient. Our differential diagnoses at primary care were whether her presentation could be due to mood disorders with features of catatonia,corticobasal neurodegeneration or stroke, viral encephalitis,neurosyphilis, or paraneoplastic syndrome. Other conditions that could mimic early CJD include aspergillosis, cryptococcal encephalitis, progressive multifocal leukoencephalopathy,endocrine disorders (e.g., Hashimoto’s encephalopathy), and heavy metal toxicity. Acquired immune deficiency syndrome(dementia, Parkinson’s dementia, and Huntington’s disease can present like late-stage CJD.
According to the World Health Organization criteria, a probable diagnosis of sporadic CJD requires that the patient has rapidly progressive cognitive impairment, and at least two of (1) myoclonus, (2) visual or cerebellar signs, (3)pyramidal or extrapyramidal signs, and (4) akinetic mutism,with at least positive result on one of the following laboratory tests: (1) a typical electroencephalogram which has periodic sharp wave complexes, during an illness of any duration or(2) a positive 14-3-3 cerebrospinal fluid assay in patients with a disease duration of less than 2 years before death, or (3)MRI high signal abnormalities in the caudate nucleus and/or putamen on diffusion-weighted imaging or fluid attenuated inversion recovery (FLAIR) and exclusion of alternative diagnoses with routine investigations. A brain biopsy or autopsy performed by standard neuropathology techniques or immunocytochemically is required for the definitive diagnosis [8]. On the basis of the WHO criteria, this patient fulfilled the diagnosis of probable sporadic CJD: with features of progressive cognitive impairment, myoclonus, cerebellar signs, extrapyramidal symptoms and signs, and akinetic mutism and supported by at least one positive result on the laboratory tests, which in this case is the typical MRI reports.Unfortunately, no cerebrospinal fluid analysis or electroencephalogram was performed to substantiate the diagnosis of sporadic CJD.
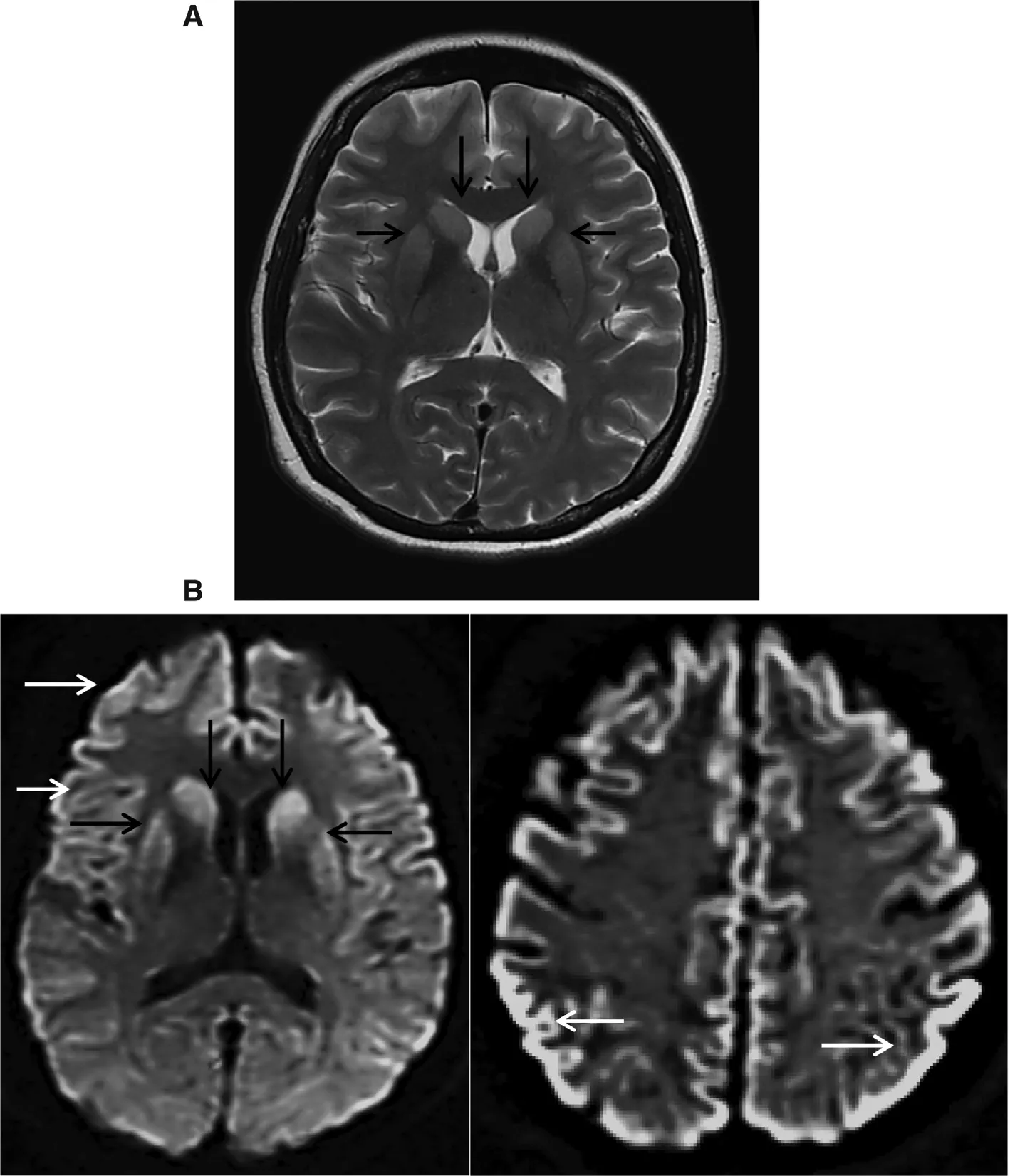
Fig. 1. MRI of brain: (A) T2-weighted imaging showing hyperintensity in bilateral caudate nuclei and putamen (black arrows); (B) diffusionweighted imaging showing homogeneous hyperintensity of bilateral caudate nuclei and putamen (black arrows) and generalized cortical ribboning hyperintensity of frontal and parietal—occipital lobes (white arrows).
In most patients, deterioration of cognition, extrapyramidal features, or cerebellar features becomes apparent early in the course of the illness [9]. Psychiatric manifestations are rare in sporadic CJD. A retrospective study by Will and Matthews[9] documented prodromal psychiatric manifestations was approximately 18%. Wall et al. [10] reported 26% of patients have psychiatric symptoms at disease onset. The atypical psychiatric features at disease onset would hinder the early diagnosis of CJD because it would not have met the existing diagnostic criteria. In primary care, it is important to make an accurate diagnosis because some of the differential diagnoses can be treated. Early recognition of CJD will allow the patient and the family members to make an informed decision on palliative management and prepare for the poor prognosis of this life-threatening disease.
Verbal consent was obtained from the patient’s husband and family members for this case presentation. The author thanks Nazifi Shkori, who treated this patient with the author in the primary care clinic, the neurology specialists, and the clinical radiologist of the University Malaya Medical Center, who was involved in the treatment of this patient while she was in the ward.
Conflict of interest
The author declares no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
1. CDC. Surveillance for Creutzfeldt-Jakob disease — United States.MMWR 1996;45(31):665—8.
2. Law ZK, Subramaniam SR, Tan HJ, Azmin S, Osman SS, Nafisah WN. Creutzfeldt-Jakob disease: a first case series from a tertiary hospital in Malaysia and review of literature in Southeast Asia.Clin Res Infect Dis 2014;1(2):1008.
3. See SJ, Pan A, Seah A, Teo J, Cham LL, Wong MC. Case reports of two biopsy-proven patients with Creutzfeldt-Jakob disease in Singapore. Ann Acad Med Singapore 2004;33:651—5.
4. Lolekha P, Rasheed A, Yotsarawat C. Creutzfeldt-Jakob disease in a tertiary care hospital in Thailand: a case series and review of the literature. J Mov Disord 2015;8:136—140.
5. Lu CJ, Sun Y, Chen SS. Incidence of Creutzfeldt-Jakob disease in Taiwan: a prospective 10-year surveillance. Eur J Epidemiol 2010;25(5):341—7.
6. Lim J-S, Kwon H-M, Jang J-W, Ju Y-R, Kim S, Park YH, et al.Characteristics of Korean patients with suspected Creutzfeldt-Jakob disease with 14-3-3 protein in cerebrospinal fluid: preliminary study of the Korean Creutzfeldt-Jakob disease active surveillance program. Prion 2015;9:136—43.
7. World Health Organization, Variant Creutzfeldt-Jakob disease.Fact sheet no180. 2012. Available from: www.who.int/mediacentre/factsheets/fs180/en/.
8. Global surveillance, diagnosis, and therapy of human transmissible spongiform encephalopathies: report of a WHO consultation February 9—11, 1998.
9. Will RG, Matthews WB. A retrospective study of Creutzfeldt-Jakob disease in England and Wales 1970—79. I: clinical features.J Neurol Neurosurg Psychiatry 1984;47(2):134—40.
10. Wall CA, Rummans TA, Aksamit AJ, Krahn LE, Pankratz VS.Psychiatric manifestations of Creutzfeldt-Jakob disease: a 25-year analysis. J Neuropsychiatry Clin Neurosci 2005;17(4):489—95.
Dr. Norita Hussein
9 March 2017;Accepted 9 May 2017
 Family Medicine and Community Health2017年4期
Family Medicine and Community Health2017年4期
- Family Medicine and Community Health的其它文章
- An automated management system for the community health service in China
- Survey and analysis of patient safety culture in a county hospital
- Additive manufacturing techniques and their biomedical applications
- Seguin Form Board as an intelligence tool for young children in an Indian urban slum
- Assessment of family physicians’ knowledge of childhood autism
- Counseling strategies for nutritional anemia by family physicians in Saudi Arabia, 2016: Implication for training