
The Influences of Basis Set Effect on the Structure, Energy and Non-covalent Interactions of the SiOSi Linkage in DFT Calculations
2014-08-24 09:36:14HUANGNanaXUJinzhouXUZheng
杭州師范大學(xué)學(xué)報(自然科學(xué)版) 2014年1期
HUANG Nana, XU Jinzhou, XU Zheng
(Key Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University,Hangzhou 311121, China)
1 Introduction
Siloxane polymers as a kind of organic-inorganic material show quite special structural and chemical properties compared to the normal organic compounds.The SiOSi linkages were considered to play the key role in determining the unique properties of siloxane polymers.It has been pointed out recently that the variation of the Si—O—Si angle in siloxane compounds can tune their basicity from highly hydrophobic system to hydrophilic system[1-3].This has great potential in the design of new siloxane materials with properties distinct from those of known silicones.Knowledge of its molecular properties including structure, chemical nature and non-bonded interactions may give an insight into the interpretation of the special properties of the materials bearing this SiOSi linkage.
Owing to the especial low linearization energy of the SiOSi angle (~0.37 kcal/mol), high precision in energy are normally needed for theoretical methods to give a reasonable description of the equilibrium structure of compounds including the SiOSi linkage.Although the effect of basis set to structural and energetic properties of disiloxane have already been tested by earlier studies[4-6], a systematic view about the convergence of the commonly available triple-ζ basis set (6-311G) and double-ζ basis set (6-31G) in combinations with various polarization space and diffuse function to disiloxane’s non-covalent binding energy is still unavailable.
As part of our ongoing interest in evaluating and comparing the chemical nature and the strength of the possible non-covalent interactions involved by compounds containing SiOSi and SiOC linkage, at the present work we firstly seek to determine to what extent will the geometric parameters and non-covalent interactions of siloxane bridge be depend on the basis sets.This may help us to choose the right combination for further works.
2 Computational Details
Calculations were carried out using Gaussian 09 suite of program[7].Disiloxane (H3SiOSiH3), which are the smallest molecules including the siloxane bridge, were selected as our model compounds.Among the three conformers of disiloxane (Figure 1), the double staggered C2vconformation (Css), which has been proved to be the most stable one compared to the staggered and eclipsed (Cse) and the doubly eclipsed (Cee) conformation,[8] was fully optimized using various combinations.The binding energy for hydrogen bond complex was calculated as in equation (1), in which EABis the energy of 1∶1 hydrogen bond complex of disiloxane (A) and water (B), EAand EBare the energy of disiloxane and water respectively.The zero-point energies (ZPE) and the basis set superposition error (BSSE)[9]for the complex have also been provided.Here, the binding energies that include ZPE correction are labeled as ΔEZPEand that include both ZPE and BSSE are labeled as ΔEZPE+BSSE.
ΔE=EAB-(EA+EB).
(1)
Fig.1 The three conformers of disiloxane
3 Results and Discussion
3.1GeometryThe most popular hybrid density functional method B3LYP was selected to evaluate the dependence of disiloxane structure to the basis set effect.This may help us to find the right basis set that can provide good compromise between the accuracy and computational cost.In the present work, the double-split-valence basis set (6-31G) and the triple-split-valence basis set (6-311G) with and without diffuse function on hydrogen and non-hydrogen atom with increasing polarization space have been tested.The specific basis sets tested were listed in Table 1.

Tab.1 Gaussian basis sets tested in the present work
As is well-known, to evaluate the performance of theoretical methods to compound containing SiOSi linkage, the key point is whether the method could give a good description of the short Si—O bond length, the large Si—O—Si bond angle and the low linearization energy of SiOSi bridge.It has been well established that the equilibrium conformation of disiloxane is bent, although the specific Si—O—Si angle predicted by different experimental methods is slightly different from each other[8, 10-12] ranging from 145° to 151.2°.The experimentally obtained Si—O—Si angle (151.2°)(10) and Si—O bond length (1.634?)(12) is provided in Figure 2 as the standard.These data have also been used as the benchmark geometric values of disiloxane by Zhang et.al.to evaluate the performance of 14 density functional to the silicaceous material system[13].In addition to that the data obtained by the widely used Dunning’s basis sets aug-cc-pVTZ and aug-cc-pVQZ were also provided for the easy comparison with previous published results about disiloxane and related compound.
Figure 2 depicted the optimized ∠SiOSi angle (A), Si—O bond distance (B) obtained at B3LYP/6-31G, 6-31+G, 6-311G, and 6-311+G with increasing polarization function.As can be seen that, upon extension of the polarization space from (d,p) to (3df,p), large perturbations in the equilibrium Si—O—Si angle and Si—O bond distance were observed for all the four sets of basis sets.Basis sets scheme such as 6-31+G(d,p), 6-311G(d,p) and 6-311+G(d,p) can’t give a good description of SiOSi angle.They tend to give a linear conformation as a minimum.Although the equilibrium SiOSi angle 151.1° predicted by the smaller basis set 6-31G(d,p) is quite close to the standard 151.2°, the predicted Si—O bond length is apparently overestimated.When polarization space is extended to (2d,p), the equilibrium SiOSi angle is obviously underestimated for all the four set of basis set schemes with a value of around 141.5°.The geometry parameters of disiloxane tend to be convergence when the polarization space is greater than (2df,p), indicating that the use of basis set with polarization space smaller than (2df,p) was not adequate.
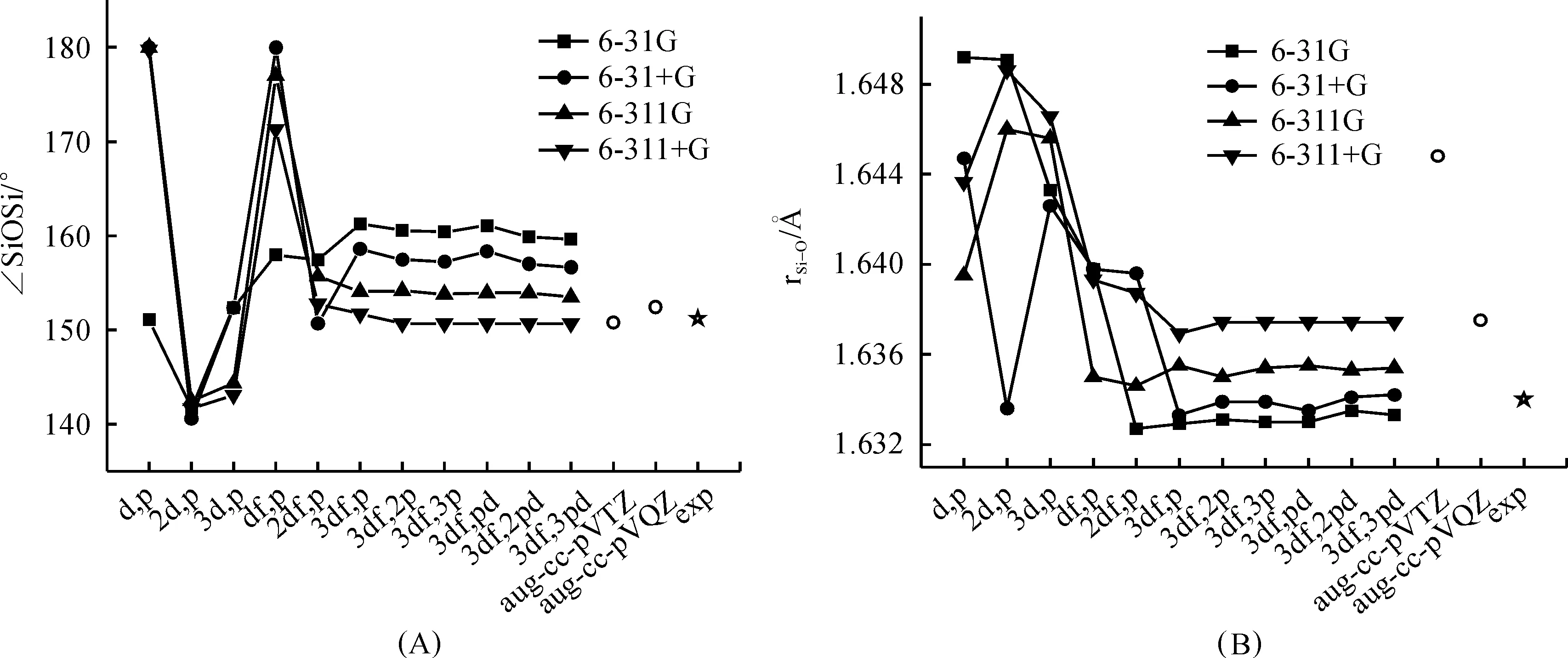
Fig.2 Optimized Si—O—Si bond angle (A) and Si—O bond distance (B) of disiloxane H3SiOSiH3 calculated at B3LYP method in conjugation with increasing basis set from 6-31G(d,p) to 6-311++G(3df,3pd) and aug-cc-pVQZ
Secondarily, it can also be seen that, after the scale of polarization space reaching the convergence point, the inclusion of diffuse function on non-hydrogen atom was found to cause a systematic decrease in SiOSi angle for about 2.8~3.1° and an increase in Si—O bond length for about 0.001~0.002?, while the inclusion of diffuse function on hydrogen atom makes nearly no difference.So the inclusion of diffuse function on non-hydrogen atom is enough.It can also be seen that the shift of basis set type from double-split-valent to triple-split-valence basis set under the same polarization space will cause a systematically decrease of 5.8~6.1° in the SiOSi angle and an increase of 0.002~0.003? in the Si—O bond length.
It has been reported that the variation of the polarization function on oxygen has only a minor influence on the molecular properties of disiloxane[14].To determine the economic basis set combination, we have tried to decrease the basis set on oxygen with the basis set for Si and H was kept to 6-311++G(3df,p) level.The influence of the polarization function on oxygen to the SiOSi angle and Si—O bond distance of the optimized disiloxane geometry have been give in Figure 3.The performance of the smallest basis set 6-31G(d,p) was found to be excellent.So in sum, basis set scheme with 6-311+G(3df,p) for Si atom and 6-31G(d,p) for oxygen atom is a good combination for predicting geometric properties of siloxane SiOSi linkage.
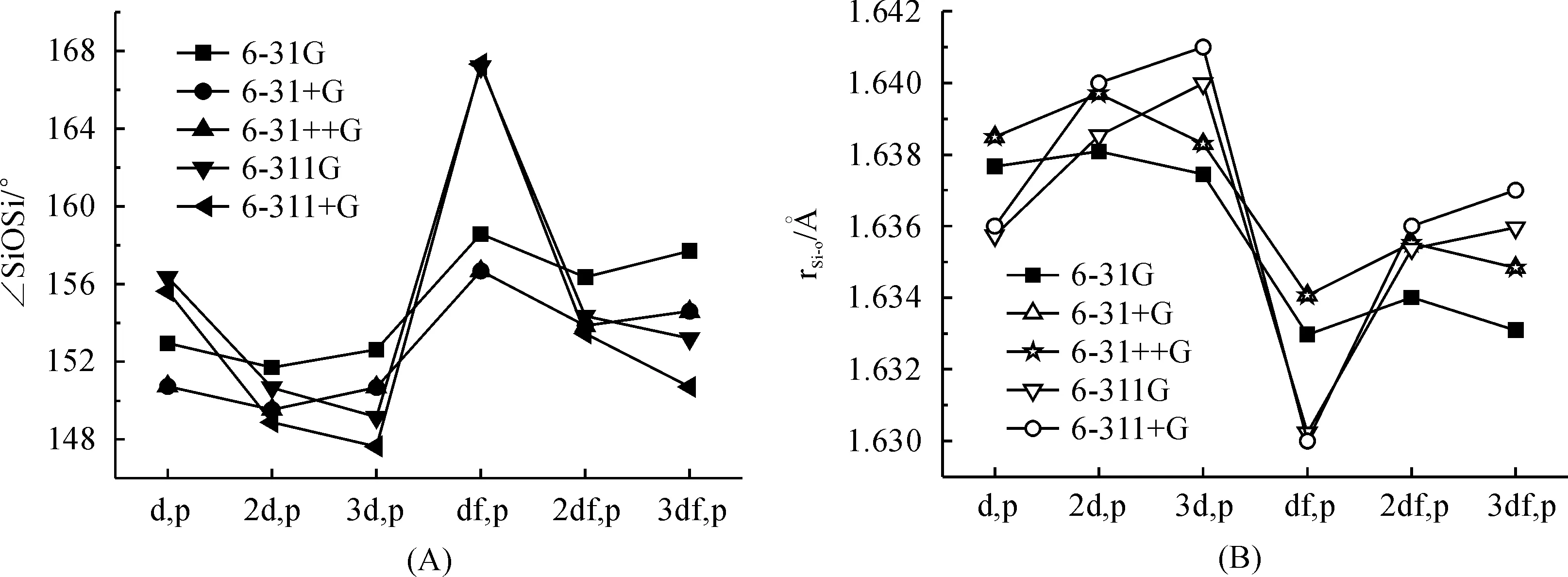
Fig.3 Effect of polarization functions in 6-31G, 6-31+G, 6-31++G, 6-311G and 6-311+G on oxygen to Si—O—Si angle (A) and Si—O bond length (B) of disiloxane calculated at B3LYP with the basis sets for Si and H atom were kept to 6-311+G(3df,p)
3.2LinearizationenergyThe sensitivity of potential energy surface of the SiOSi angle to the polarization space has been tested by a relaxed potential energy surface scan of H3SiOSiH3with the SiOSi angle was fixed between 120° and 180° within 5° intervals under B3LYP/6-311+G scheme.The PES calculated with the B3LYP/6-311+G scheme in combination with various polarization spaces are shown in Figure 4 (A).The barrier for linearization of the SiOSi group was found to correlate with the equilibrium SiOSi angle.For basis sets, such as 6-311+G(2d,p) and 6-311+G(3d,p), that predict low SiOSi angle (~141°), the linearization barrier will be slightly higher (about 0.9 kcal/mol), however the deformation energy for SiOSi angle from equilibrium structure to smaller angle such as 120° is relatively low (about 2 kcal/mol).For basis sets that predict relative large equilibrium SiOSi angle, such as 6-311+G(d,p) and 6-311+G(df,p), the deformation energy for SiOSi angle from equilibrium to 120° is 6 kcal/mol and 4 kcal/mol, which is apparently greater than that obtained with aug-cc-pVQZ (3 kcal/mol).The result indicates that the flexibility of siloxane predicted by these basis set is greatly decreased.The potential energy surface of SiOSi angle predicted by basis sets including 6-311+G(2df,p), 6-311+G(3df,p) and 6-311+G(3df,3p) is quite close that obtained by aug-cc-pVQZ.Thus choose the right basis set is very important to give a reasonable description of the flexibility of siloxane compounds and 6-311+G(3df,p) is a good candidate basis set for siloxane system.
The effect of diffuse functions on non hydrogen heavy atom (oxygen and silicon atoms) in both the double-split-valence and triple-split-valence basis set to the PES of the SiOSi angle has also been tested with the polarization space was fixed at (3df,p) and the results were shown in Figure 4 (B).It can be seen that inclusion of diffuse function on non-hydrogen bond will lead to a slightly decrease within 0.5 kcal/mol while the ∠SiOSi is narrowed from the equilibrium point to 120°, while the effect of the diffuse function on hydrogen atom is completely neglectable.
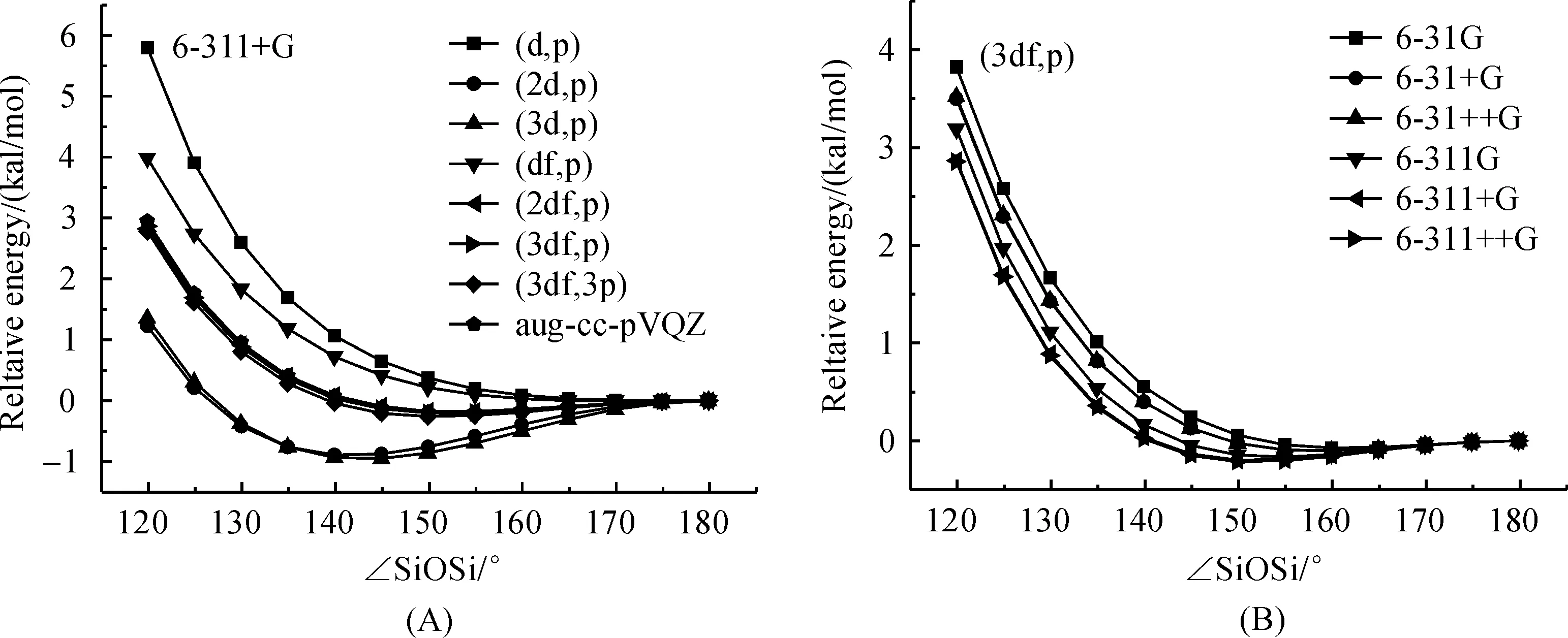
Fig.4 Effect of polarization functions (A) and diffuse function (B) on the Si—O—Si bending potential energy surface of disiloxane H3SiOSiH3 calculated at B3LYP in combination with different basis set scheme
In addition to that the effect of decreasing polarization functions on oxygen to the Si—O—Si bending potential energy surface have also been tested using B3LYP method with the basis sets for Si and H atom were kept to 6-311+G(3df,p) and the results were shown in Figure 5.The polarize space (d,p) and (2df,p) were found to give PES quite similar to that obtained by the (3df,p) space.Taking result of (3df,p) as standard, the PES predicted by (df,p) is higher and that by (2d,p) and (3d,p) is lower than the standard.
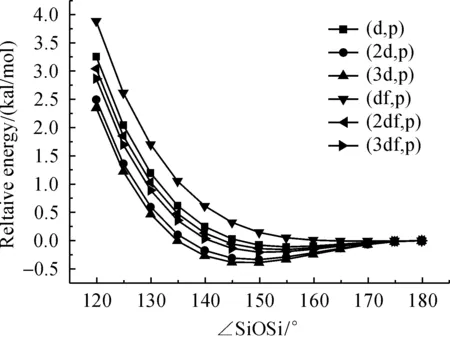
Fig.5 Effect of polarization functions in 6-311+G on oxygen to the Si—O—Si bending potential energy surface of disiloxane H3SiOSiH3 calculated at B3LYP method with the basis sets for Si and H atom were kept to 6-311+G(3df,p)
3.3Non-covalentinteractionsThe 1∶1 complex (H3Si)2O…HOH was selected to test the weak hydrogen bonds involved by oxygen in disiloxane, where H2O acted as hydrogen bond donor.Although the nonlocal exchange-correlation functional B3LYP can give good results in predicting the structural and energetic properties of organic compounds, its performance in evaluating the weak interactions between hydrocarbon molecules was very poor[15].The PBE1PBE method, which has excellent performance in hydrogen bonds and weak interactions, was thus selected as the model method[16-17].The binding energies and hydrogen bond length for 1∶1 complex between H3SiOSiH3and H2O optimized at PBE1PBE method with 6-311++G in combination with various increasing polarization space is depicted in Figure 6.The distance between the oxygen atom of H3SiOSiH3and hydrogen atom of water were fully optimized without any constraints.It can be seen that binding energy and hydrogen bond length achieve convergence at 6-311+G(3df,p) with -1.25 kcal/mol.The binding energy predicted by basis set with small polarization space such as (d,p), (2d,p) and (df,p) tend to be overestimated by 0.4~0.8 kcal/mol.
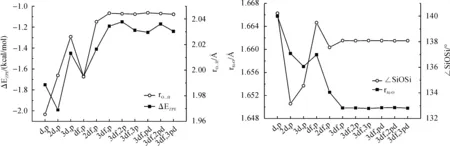
Fig.6 Binding energy with zero-point correction (ΔEZPE) and hydrogen bond length for H3SiOSiH3-H2O 1∶1 complex calculated at PBE1PBE method with 6-311+G in combination with various increasing polarization space
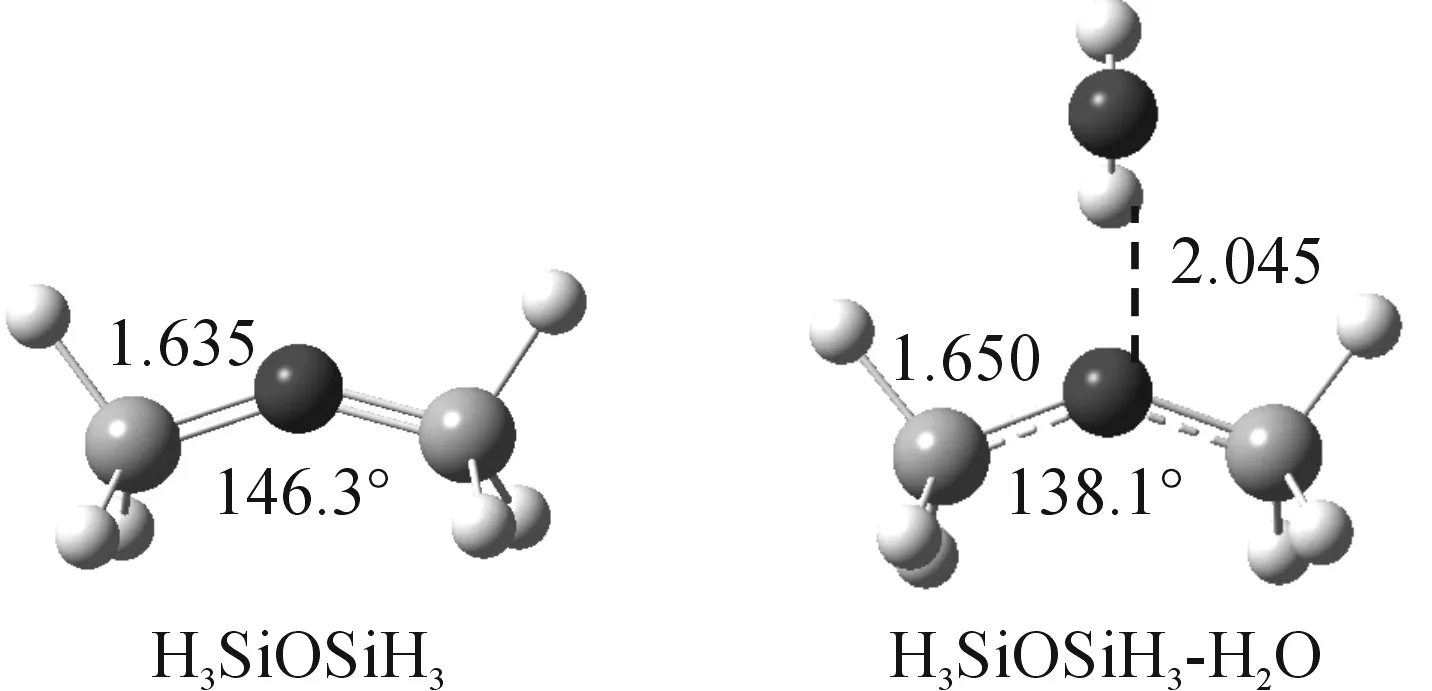
Fig.7 Optimized structure of disiloxane and its 1∶1 complex with water calculated at PBE1PBE/6-311+G(3df,p) level of theory
The optimized geometry of disiloxane monomer and the hydrogen bond complex calculated at PBE1PBE/6-311+G(3df,p) was given in Figure 7.Compared to the disiloxane monomer, the Si—O bond is elongated by 0.027? and the SiOSi angle is narrowed by 8.2° in the hydrogen bond complex.The results suggest that the participation of the oxygen atom to hydrogen bond is likely to cause electronic structure change of the SiOSi linkage.
Relaxed potential energy surface scan for H3SiOSiH3-H2O 1∶1 complex were carried out at PBE1PBE/6-311+G(3df,p) level of theory with the SiOSi angle was fixed between 120° and 180° within 5° intervals.The corresponding binding energy (ΔE) of H3SiOSiH3-H2O 1∶1 complex including ZPE and ZPE+BSSE has been shown in Figure 8 A.When SiOSi angel was greater than 170°, no stationary point for the hydrogen bond complex were found.Interestingly, a gradual increase in the binding energy was observed from 0.0 kcal/mol to 1.6 kcal/mol when the SiOSi angle was narrowed from 170° to 120°.This is accompanied by a decrease of hydrogen bond length RO…Hdefined as the intermolecular distance between disiloxane oxygen and water hydrogen atom that is pointing to disiloxane oxygen atom.This is in consistent with the results of Grabowsky et.al.that H3SiOSiH3can form usual hydrogen bond with proton donor such as H3SiOH and H2O under small SiOSi angle.(1) Additionally, the inclusion of ZPE effect was found to cause a systematic decrease in binding energy for about 1.3 kcal/mol while the inclusion of BSSE effect is about 0.35 kcal/mol.The results indicate that ZPE is important to give a reasonable description of non-covalent interactions involved by siloxane compounds.
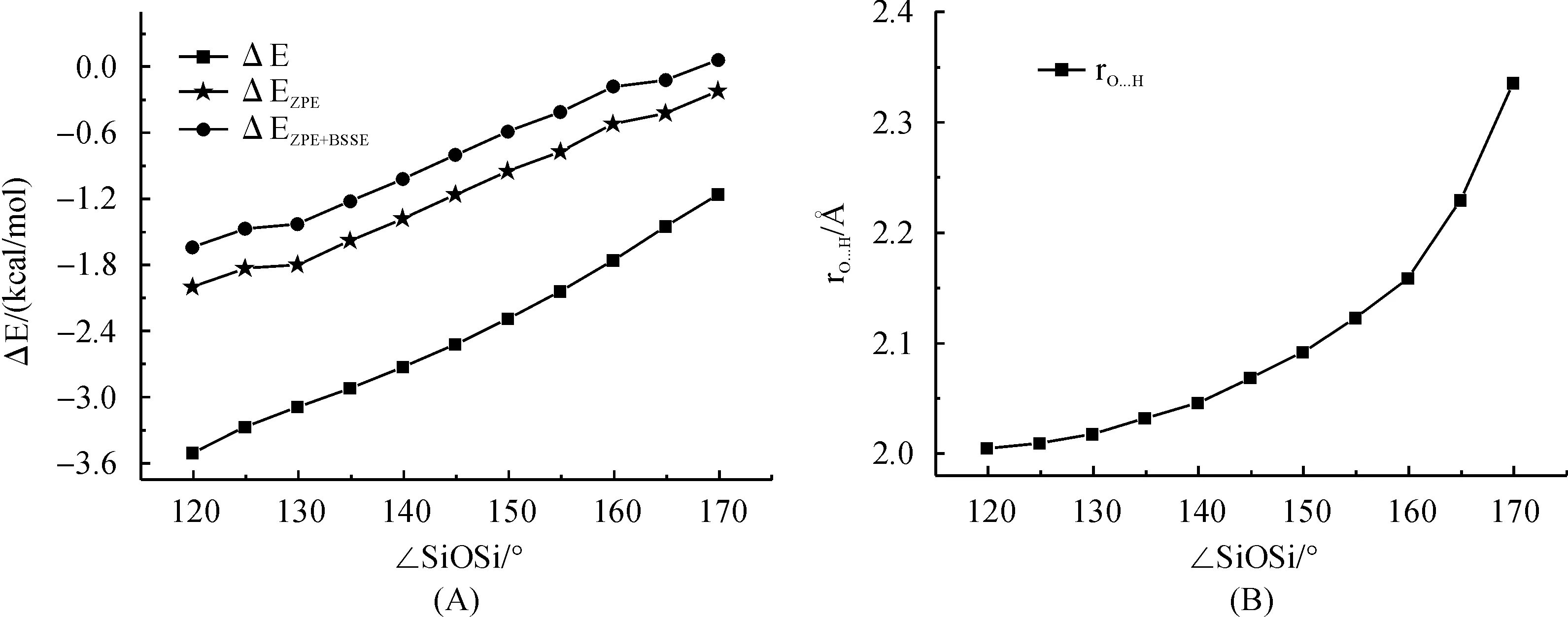
Fig.8 Binding energy (including ΔE, ΔEZPE and ΔEZPE+BSSE) and hydrogen bond length (RO…H in ?) for H3SiOSiH3-H2O 1∶1 complex predicted by PBE1PBE/6-311+G(3df,p) method with the ∠SiOSi angle are fixed at different value between 120°-180°
4 Conclusions
A systematic analysis of the effects of the basis sets to the geometry, linearization barrier and weak non-bonded interaction of siloxane model compound disiloxane have been carried out.Basis set scheme 6-311+G(3df,p)/6-31G(d,p) (the first basis is for Si atom and the last set if for all other atoms) was found to be a good basis set scheme that can give the best compromise between accuracy and CPU time for both geometrical characterization and no-covalent interactions of the SiOSi linkage in siloxane.
The inclusion of ZPE is needed to give a reasonable description of non-covalent binding energy while basis set superposition error (BSSE) is however neglectable.The PES of SiOSi angle is sensitive to the polarization space, while the effect of diffuse function on both hydrogen and non-hydrogen atom is small.
[1] Grabowsky S, Beckmann J, Luger P.The nature of hydrogen bonding involving the siloxane group[J].Aust J Chem, 2012, 65(7):785-795.
[2] Weinhold F, West R.The nature of the silicon-oxygen bond[J].Organometallics,2011, 30(21):5815-5824.
[3] Grabowsky S, Hesse M F, Paulmann C,etal.How to make the ionic Si—O bond more covalent and the Si—O—Si linkage a better acceptor for hydrogen bonding[J].Inorg Chem,2009, 48(10):4384-4393.
[4] B?r M R, Sauer J.Ab initio calculations of the structure and properties of disiloxane.The effect of electron correlation and basis set extension[J].Chem Phys Lett,1994,226(3):405-412.
[5] Nicholas J B, Winans R E, Harrison R J,etal.An ab initio investigation of disiloxane using extended basis sets and electron correlation[J].J Phys Chem,1992,96(20):7958-7965.
[6] Nicholas J B, Winans R E, Harrison R J,etal.Ab initio molecular orbital study of the effects of basis set size on the calculated structure and acidity of hydroxyl groups in framework molecular sieves[J].J Phys Chem,1992,96(25):10247-10257.
[7] Frisch M, Trucks G W, Schlegel H B,etal.Gaussian 09, Revision A.02[CP].Gaussian Inc, Wallingford, CT 2009,270:271.
[8] Carteret C, Labrosse A, Assfeld X.An ab initio and DFT study of structure and vibrational spectra of disiloxane H3SiOSiH3conformers-Comparison to experimental data[J].Spectrochim Acta,Part A,2007,67(5):1421-1429.
[9] Boys S F, Bernardi F.Calculation of small molecular interactions by differences of separate total energies-some procedures with reduced errors[J].Mol Phys,1970,19(4):553-566.
[10] Almenningen A, Traetteberg M, Hedberg K,etal.Molecular structure of disiloxane(SIH3)2O[J].Acta Crystallogr, Sect.B:Struct Sci,1963,17(9):2455-2460.
[11] Durig J R, Flanagan M J, Kalasinsky V F.Determination of potential function governing low-frequency bending mode of disiloxane[J].J Chem Phys,1977,66(7):2775-2785.
[12] Koput J, Wierzbicki A.The large-amplitued motions in quasi-symmetric top molecules with internal C3V rotors-interpretation of the low-frequency raman-spectrum of disiloxane[J].J Mol Spectrosc,1983,99(1):116-132.
[13] Zhang Y, Li Z H, Truhlar D G.Computational requirements for simulating the structures and proton activity of silicaceous materials[J].J Chem Theory Comput,2007,3(2):593-604.
[14] Grigoras S, Lane T H.Ab initio calculations of the effect of polarization functions on disiloxane[J].J Comput Chem,1987,8(1):84-93.
[15] Sousa S F, Fernandes P A, Ramos M J.General performance of density functionals[J].J Phys Chem A,2007,111(42):10439-10452.
[16] Rabuck A D, Scuseria G E.Performance of recently developed kinetic energy density functionals for the calculation of hydrogen binding strengths and hydrogen-bonded structures[J].Theor Chem Acc,2000,104(6):439-444.
[17] Ireta J, Neugebauer J, Scheffler M.On the accuracy of DFT for describing hydrogen bonds:dependence on the bond directionality[J].J Phys Chem A,2004,108(26):5692-5698.
