
FDA Practices: GMP Quality System Inspection and Evaluation
2014-05-15 07:56LIYudanYANGYue
亞洲社會藥學(xué)雜志 2014年4期
LI Yu-dan, YANG Yue
(School of Business Administration, Shenyang Pharmaceutical University, Shenyang 110016, China)
FDA Practices: GMP Quality System Inspection and Evaluation
LI Yu-dan, YANG Yue
(School of Business Administration, Shenyang Pharmaceutical University, Shenyang 110016, China)
Objective To regulate and guide pharmaceutical manufacturers to set up a better quality system and to ensure drug safety by studying FDA practices with ICH Q10 and 483 warning letters. Methods FDA practices at the inspection and evaluation were studied through literature review and statistical analysis methods. Results and Conclusion FDA attaches importance to the construction of inspection team during the evaluation process and focuses on the establishment of written procedures and process control with corrective and preventive measures.
quality system; ICH Q10; inspection
China’s new GMP came into force on March 1st, 2011. New concepts, such as quality system and risk management being introduced into the new GMP showed a big step forward towards the advanced international standard. However, after the implementation of GMP, how to inspect and evaluate the quality system built by factories remains a problem which needs to be settled urgently. It is a wise approach to draw from FDA’s rich experience that has done the inspection of pharmaceutical manufacturers based on quality system and risk management for over a decade. To safeguard drug safety and prevent the adverse drug events from happening, the practices introduced in this article has practical significance on guiding the inspection and evaluation of quality system.
1 Quality system
1.1 Introduction of quality system
As early as in August 2002, FDA announced a significant new initiative, Pharmaceutical cGMPs for the 21st Century, A Risk-based Approach, to enhance and modernize the regulation of pharmaceutical manufacturing and product quality which brings together the quality system and risk management[1]. Though FDA remained the 210 and 211 parts of 21 CFR (Code of Federal Regulations, CFR) unchanged, FDA issued a industry guidance “Quality Systems Approach to Pharmaceutical cGMP Regulations”in September, 2006 to guide pharmaceutical manufacturer to build quality system in compliance with cGMP[2].
Afterwards, the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) established an ICH tripartite guideline describing a model referred to as the Pharmaceutical Quality System, Q10. Q10 is based on International Standards Organization (ISO) quality concepts, includes applicable GMP regulations and complements ICH Q8 “Pharmaceutical Development” and ICH Q9“Quality Risk Management”. ICH Q10 is a model for a pharmaceutical quality system that can be implemented throughout the different stages of a product lifecycle.
FDA and ICH Q10 share the same concept on quality system, while based on FDA’s quality system, Q10 has improved the quality system to make it more concrete and systematic. This article will follow the terminology of Q10.
1.2 Key elements of pharmaceutical quality system
Pharmaceutical quality system contains four key elements:
a. Process performance and product quality monitoring system;
b. Corrective action and preventive action (CAPA) system;
c. Change management system;
d. Management review of process performance and product quality.
In process performance and product quality monitoring system, pharmaceutical companies should plan and execute a system for the monitoring of process performance and product quality to ensure a state of control is maintained. An effective monitoring system provides assurance of the continued capability of processes and controls to produce a product of desired quality and to identify areas for continual improvement.
In CAPA system, the pharmaceutical company is required to have a system for implementing corrective actions and preventive actions resulting from the investigation of complaints, product rejections, nonconformances, recalls, deviations, audits, regulatory inspections and findings and trends from process performance and product quality monitoring. A structured approach to the investigation process should be used with the objective of determining the root cause.
Innovation, continual improvement, the outputs of process performance and product quality monitoring and CAPA drive change. In order to evaluate, approve and implement these changes properly, a company should have an effective change management system.
Management review should provide assurance that process performance and product quality are managed over the lifecycle. Depending on the size and complexity of the company, management review can be a series of reviews at various levels of management and should include a timely and effective communication and escalation process to raise appropriate quality issues to senior levels of management for review[3].
2 Inspection of quality system
2.1 Change of inspection concept
FDA’s inspection concept has been changing with the development of GMP. A flexible GMP inevitably leads to up-to-date GMP inspections. Focusing on ex-factory inspection cannot guarantee the product consistency and is laggard and hysteretic which already doesn’t meet the cGMP inspection requirements. With the improvement of pharmaceutical regulation, the concepts of process control and “Quality by Design” are recognized world-wide. All those have put forward higher requirement for inspection. The investigator should pay close attention to the whole situation of a factory, especially its quality system. As long as the quality system keeps in a controlled state, the drugs manufactured are definitely qualified even without inspection.
2.2 Construction of inspection team
Not only did FDA apply the quality system to guiding pharmaceutical manufacturers, but also to the construction of inspection team. “Quality System” here doesn’t refer to product quality system, but the inspection quality system.“Quality” refers to all the regulation and activities carried out by FDA. The Quality System Framework Working Group formed by FDA developed a model for the cGMP initiative, referencing key recognized external quality and risk management standards. This quality system model, now incorporated into the FDA Staff Manual Guide, Quality System Framework for Internal Activities[4]will guide FDA’s inspection and other regulatory activities.
2.3 Upgrading of inspection approach- six-system based inspection
On February 1st, FDA issued “Compliance Program Guidance Manual Program 7356.002”[5]introducing the procedures and methods of inspection. It requires that inspections of drug manufacturers should be made and reported using the system definitions and organization in this compliance program. Inspection is carried out based on the following six systems: Quality System, Facilities and Equipment System, Materials System, Production System, Packaging and Labeling System and Laboratory Control System.
“Quality Systems Approach to Pharmaceutical cGMP Regulations” announced by FDA in September, 2006 describes in detail how the pharmaceutical manufacturer can build quality system that meets the requirement of cGMP. It emphasizes again FDA will use the systematic approach, i.e. six-system based inspection. The relationship between the six systems is that quality system embodies the other five systems while they overlap each other.
3 Evaluation of quality system
3.1 Revision of regulatory procedure
It is notable that FDA does not have details on how to evaluate quality system. It is the investigator’s rich experience the inspection is counted on. Through tiny deficiencies, the investigator can predict the potential discrepancy of the whole quality system. GMP investigator is only responsible for describing the violations and bringing these issues back to office, but he or she will not make the decision of whether the factory is passing the GMPinspection or not. After the inspection, the investigator will write down all the violations observed on the Form 483, also called FDA 483 or 483, which is used by FDA to document and communicate concerns discovered during these inspections and provide it to the manager as a notification of violations against GMP. Back to the office, the investigator will submit the violations to the inspection team who will make the decision of whether to issue the warning letter or not[6]. Based on the response to the warning letter, they will determine whether the company passes the cGMP or not. A warning letter is an official notice to a regulated business establishment that objectionable conditions or practices have been identified in their operations, that corrections are expected, and that failure to correct the deficiencies may result in further FDA actions. Therefore, the issuance of warning letter is closely related to the pass of cGMP.
In the Second Progress Report and Implementation Plan of Pharmaceutical cGMPs for the 21st Century —A Risk-Based Approach, FDA has revised the regulatory procedure it uses for determining when to issue warning letters in response to noncompliance with cGMP requirements. Since March 1st, 2003, all proposals to issue a warning letter have been reviewed by the centers with product jurisdiction. The appropriate center will determine whether the letter can be issued. A single office authority for the approval of all letters will ensure that requirements are applied consistently. Incorporating a center role in the process will ensure that adverse findings will be based on the best science available and will enhance communication and coordination between the fields and centers and help identify possible program inconsistencies for resolution before the issuance of a warning letter[7].
3.2 Focus of evaluation and trend analysis
Analysis of deficiencies on FDA 483 and warning letters will help us understand the focus and trend of FDA’s evaluation. The statistical summary of FDA 483 and analysis of warning letter will display the common deficiencies during inspection and show the trend and focus of FDA’s evaluation of quality system.
3.2.1 Emphasizing on written procedures
In most cases, FDA will not publish the FDA 483 of a certain company; however, FDA will announce the annual total statistics. Table 1 illustrates a list of top 10 deficiencies sorted in line with the key elements of quality system during the inspection from September 2005 to September 2011. Notably, most of the deficiencies belong to process performance and product quality monitoring system, while the major problem is failure to establish or follow written procedures.
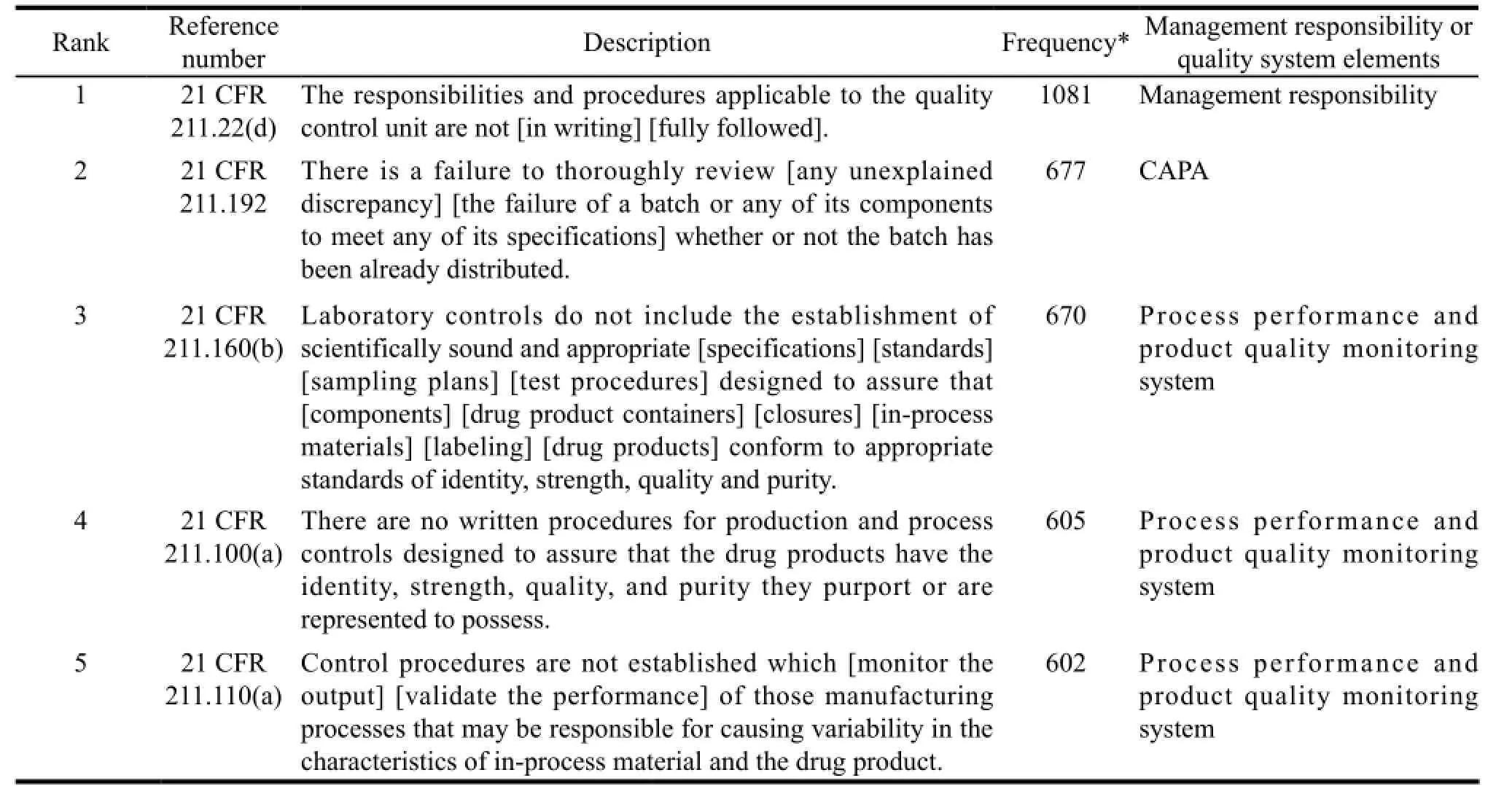
Table 1 Top 10 deficiencies of FDA 483 from September 2005 to September 2011[8]
For any of the six systems, lacking written procedures is considered as significant deficiency. Besides, Douglas A. Campbell, a compliance officer, once remarked in his speech that for old FDA, there is a mantra: “If you didn’t document it, then it didn’t happen…”[9].
3.2.2 Enough CAPA
FDA requires a thorough investigation of the discrepancies found in the process of manufacture and detection. An analysis of FDA warning letters will provide us an insight into the requirements of CAPA. During June 17th, 2013 through June 27th, 2013 inspection of a pharmaceutical manufacturing facility, Agila Specialties Private Limited, investigators from FDA identified significant violations of cGMP regulations[10]. Table 2 describes a warning letter which constitutes a typical example of the extent of CAPA FAD is expecting.
Apparently, FDA is not satisfied with company’s solution to discontinue the glove vendor. Although the solution may correct the deficiencies found, it doesn’t find the root cause of the problem and cannot prevent similar deficiencies from happening. Company’s reply indicating that the gloves all meet the requirements leads to FDA’s doubt about company’s vendor qualification program. Meanwhile, the gloves are approved and released by QA which bring FDA’s concern about the incoming material release system. Therefore, FDA didn’t agree that the company is in a state of control.
3.2.3 Quality system focused
FDA’s decision to the pass of GMP depends on the extent of quality system built by the company instead of the inspection result. Although products are tested to be qualified, FDA doesn’t allow an unreliable quality system. FDA focuses more on the process control rather than inspection results. FDA’s inspection relies on data. If the data is forged, FDA will doubt everything of the company. For example, Ranbaxy, an Indian pharmaceutical company, forged the data and FDA said Ranbaxy won’t be allowed to make drugs for the U.S. until the company comes into compliance with FDA manufacturing rules. Notably, in the press conference, FDA repeatedly emphasized that all the product tests show all the products are qualified and have no safety issue. This demonstrates that FDA is quality system focused rather than product focused.
4 Revelation
The concept of quality system is widely recognized and introduced into GMPs in various countries, including China. China Food and Drug Administration (CFDA) issued “Measures for Drug GMP Certification” and“Risk Management Principals of Field Inspection of Pharmaceutical Company” indicating a further step forward on the regulatory level. In reality, we should draw from FDA’s experience, particularly, the construction of inspection team to collaborate with the implementation of quality system, the focused trend and parts during inspection and the emphasizing on CAPA. The concepts and practices are worth learning so as to better safeguard drug safety and reduce adverse drug events.
[1] FDA. Pharmaceutical cGMPs for the 21st Century - A Risk-Based Approach [EB/OL]. http://www.fda.gov/drugs/developmentappro valprocess/manufacturing/questionsandanswersoncurrentgoodma nufacturingpracticescgmpfordrugs/ucm137175.htm.
[2] FDA. Guidance for Industry Quality Systems Approach to Pharmaceutical cGMP Regulations [EB/OL]. http://www.fda.gov/ downloads/Drugs/.../Guidances/UCM070337.pdf.
[3] FDA. Guidance for Industry Q10 Pharmaceutical Quality System [EB/OL]. http://www.fda.gov/downloads/Drugs/Guidances/ ucm073517.pdf.
[4] FDA. FDA Staff Manual Guides, Volume Iii - General Administration [EB/OL]. http://www.fda.gov/AboutFDA/ ReportsManualsForms/StaffManualGuides/ucm052570.htm.
[5] FDA. Compliance Program Guidance Manual Program [EB/OL]. http://www.fda.gov/downloads/ICECI/ComplianceManuals/ ComplianceProgramManual/ucm125404.pdf.
[6] FDA. Inspections, Compliance, Enforcement and Criminal Investigations [EB/OL]. http://www.fda.gov/ICECI/ ComplianceManuals/RegulatoryProceduresManual/ucm176870. htm#SUB4-1-4.
[7] FDA. Pharmaceutical cGMPs for the 21st Century — A Riskbased Approach: Second Progress Report and Implementation Plan Introduction [EB/OL]. http://www.fda.gov/Drugs/Developm entApprovalProcess/Manufacturing/QuestionsandAnswersonCurr entGoodManufacturingPracticescGMPforDrugs/UCM071836.
[8] FDA. Inspections, Compliance, Enforcement and Criminal Investigations [EB/OL]. http://www.fda.gov/ICECI/ EnforcementActions/ucm255532.htm.
[9] FDA. Conducting the FDA Inspection [EB/OL]. http://www.fda. gov/downloads/Drugs/NewsEvents/UCM167322.pdf.
[10] FDA. Inspections, Compliance, Enforcement and Criminal Investigations [EB/OL]. http://www.fda.gov/ICECI/ EnforcementActions/WarningLetters/2013/ucm369407.htm.
Author’s information: YANG Yue, Professor. Major research area: Pharmacy administration. Tel: 024-23986372, E-mail: yyue315@126. com

- 亞洲社會藥學(xué)雜志的其它文章
- Marine Pharmaceutical Industry Development in Developed Countries and Its Enlightenment
- Study on the Establishment of Efficiency Evaluation System of Drug Supervision and Sampling Test
- New Version of GDP in EU and Its Enlightenment
- Family Economic Risk Caused by Five Major Chronic Diseases among Urban Residents: A Comparative Study Based on the Data from 9 Cities in China
- Three Therapeutic Regimens in Treatment of Community-acquired Pneumonia: A Cost-effectiveness Analysis
- Pharmacovigilance System for Pharmaceutical Enterprises in EU: Regulation and Its Enlightenment