
Dissecting the key genomic regions underlying high yield potential in common wheat variety ‘Kenong 9204’
2023-09-16 02:36:50ZHAOChunhuaZHANGNaFANXiaoliJlJunSHlXiaoliCUlFaLlNGHongqingLlJunming
ZHAO Chun-hua,ZHANG Na,FAN Xiao-li,Jl Jun ,SHl Xiao-liCUl Fa,LlNG Hong-qing#,Ll Jun-ming,
1 College of Agriculture,Ludong University/Key Laboratory of Molecular Module-based Breeding of High Yield and Abiotic Resistant Plants in Universities of Shandong,Yantai 264025,P.R.China
2 Jiangsu Xuhuai Regional Institute of Agricultural Sciences,Xuzhou 221131,P.R.China
3 Chengdu Institute of Biology,Chinese Academy of Sciences,Chengdu 610041,P.R.China
4 State Key Laboratory of Plant Cell and Chromosome Engineering,Institute of Genetics and Developmental Biology,Innovation Academy for Seed Design,Chinese Academy of Sciences,Beijng 100101,P.R.China
5 Center for Agricultural Resources Research,Institute of Genetics and Developmental Biology,Chinese Academy of Sciences,Shijiazhuang 050022,P.R.China
6 Key Laboratory of Molecular and Cellular Biology,Ministry of Education/Hebei Collaboration Innovation Center for Cell Signaling/Hebei Key Laboratory of Molecular and Cellular Biology/College of Life Sciences,Hebei Normal University,Shijiazhuang 050024,P.R.China
Abstract
The foundation parents play key roles in the genetic improvement of both yield potential and end-use quality in wheat.Characterizing the genetic basis that underlies certain beneficial traits in the foundation parents will provide theoretical reference for molecular breeding by a design approach.‘Kenong 9204’ (KN9204) is a candidate foundation parent characterized by ideotype,high yield potential,and particularly high nitrogen fertilizer utilization.To better understand the genetic basis of its high yield potential,high throughput whole-genome re-sequencing (10×) was performed on KN9204,its parental lines and its derivatives.A high-resolution genetic composition map of KN9204 was constructed,which showed the parental origin of the favorable genomic segments based on the identification of excellent yield-related quantitative trait loci (QTL) from a bi-parental mapping population.Xiaoyan 693 (XY693),a wheat–Thinopyrum ponticum partial amphidiploid,contributed a great deal to the high yield potential of KN9204,and three major stable QTLs from XY693 were fine mapped.The transmissibility of key genomic segments from KN9204 to its derivatives were delineated,indicating that haplotype blocks containing beneficial gene combinations were conserved along with directional selection by breeders.Evidence for selection sweeps in the breeding programs was identified.This study provides a theoretical reference for the breeding of high-yield wheat varieties by a molecular design approach.
Keywords: Kenong 9204,high yield potential,quantitative trait locus,genetic composition map,key genomic regions
1.lntroduction
Wheat (TriticumaestivumL.) is a crop of global importance that accounts for approximately 20% of the total dietary calories and protein consumed worldwide (Shiferawetal.2013; Mohammad and Donald 2019).Phenotype-based selection in traditional breeding programs has resulted in the great phenotypic changes eventually seen in modern crops (Doebleyetal.2006; Meyeretal.2012).In traditional breeding programs,approximately 1.0% of a breeder’s crosses result in the release of a commercial variety (Zhuang 2003),and result in annual genetic gains of approximately 1.0% in the five major food crops (Rayetal.2013).However,limited knowledge about the genetic basis of the parental lines,especially the most widely used key parents,might be the cause of the low breeding efficiency observed in breeding programs today.
Foundation parents or genotypes refer to the breeding materials that not only give rise to many varieties (lines) but also occupy a large planting area during a certain period,and they have played important roles in the genetic improvement of wheat (Zhuang 2003).Triticeae consists of over 30 genera and 350 species (Lietal.2021),and provides many important gene pools in the process of breeding wheat foundation parents,with the 1BL/1RS wheat–rye translocation line as a typical case (Friebeetal.1987; Rabinovichetal.1998).In addition,Thinopyrumhas received more attention by both wheat breeders and geneticist,since it has contributed several valuable disease resistance genes to wheat (Shenetal.2007; Li and Wang 2009; Ceolonietal.2017).Fhb7,which was isolated recently,confers resistance to both Fusarium head blight (FHB) and crown rot in diverse wheat backgrounds without a yield penalty (Wangetal.2020).Introgressions ofThinopyrumponticumchromosomal segments were shown to contain multiple beneficial gene(s)/QTLs for yieldrelated traits and salt tolerance (Kuzmanovi?aetal.2016; Tongetal.2022; Yangetal.2022).So it is significant to characterize the genetic effects of alien chromosomal segments in foundation parents.
The advancement in molecular marker analysis technologies along with the genomic sequencing technologies currently allow for a more objective appraisal of the necessary features for becoming a foundation parent or founder genotype (Shietal.2022).The breeding process has targeted some genomic regions,i.e.,selective sweeps,of foundation parents that probably contain the excellent alleles relevant to the most critical agronomic traits.Through the combination of pedigree analysis and molecular marker screening,the selection effects of key genomic regions on elite varieties,including foundation parents,have been reported in many studies (Lorenzenetal.1995; Hanetal.2009; Sietal.2009; Zhaoetal.2013; Dengetal.2018; Gaireetal.2020; Haoetal.2020).Identifying the key genomic regions not only enables our understanding of the regular pattern of foundation parent formation,but also guides our practice in breeding new wheat varieties with significantly improved traits by pyramiding the elite alleles through genome-wide marker assisted selection.The 5G breeding approach is a brand new concept in molecular breeding.The 1st G is Genome assembly for each crop species,the 2nd G is Germplasm characterized at the genomic and agronomic levels,the 3rd G is Gene function identification,the 4th G is Genomic breeding methodologies,and the 5th G is Gene editing technologies.The comprehensively applied 5G breeding approach can enhance the precision,efficiency and effectiveness of breeding programs (Varshneyetal.2019).Genome sequence information along with the key gene characterization are essential for achieving the 5G breeding approach.It is critical to have a deep understanding of the genetic effects of key genomic regions in elite germplasms as well as the foundation parents.Advances in sequencing technologies along with the Chinese Spring reference genome have facilitated the re-sequencing of some important wheat germplasms (https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Assemblies/v1.0/) (Chengetal.2019).In general,three steps should be followed to comprehensively understand the genetic basis of a foundation parent.Firstly,the genotypic map of the foundation parents along with its derived varieties should be released; secondly,key genomic regions with hitchhiking effects should be recognized; and lastly,the genetic effects of key genomic regions should be characterized,and the key genes along with functional markers should be specified,if possible.
‘Kenong 9204’ (KN9204) is characterized by a semidwarf plant type,erect flag leaf,high yield potential,convenient disease resistance,a larger root system and particularly high nitrogen use efficiency (NUE) (Cuietal.2011,2016; Wangetal.2011; Fanetal.2018).It has been used extensively in wheat breeding programs,and dozens of advanced varieties derived from KN9204 have been authorized,including ‘Kenong 1093’ (registration number,JS2005005),‘Kenong 199’ (registration number,GS2006017),‘Kenong 2011’ (registration number,JS2016003),‘Kenong 2009’ (registration number,GS20170015),and others.Recently,we completed the genome assembly of KN9204 and its genome annotation in order to understand its genetic basis of high yield potential and NUE in greater detail (Shietal.2022).
We released a high-resolution genetic composition map of KN9204 based on the high throughput wholegenome re-sequencing in this study.Combined with quantitative genetic analysis,the selection effects of key genomic segments from KN9204 to its derivatives were characterized.The primary goals of this study were: 1) to dissect the genetic composition of the candidate foundation parent KN9204; 2) to identify the genetic factors underlying high grain yield potential in KN9204; 3) to specify the key genomic regions with hitchhiking effects in KN9204; and 4) to fine map some major stable quantitative trait loci (QTLs) from the wheat–Th.ponticumpartial amphiploid XY693.
2.Materials and methods
2.1.Plant materials,trait evaluation and QTL detection
The common wheat variety KN9204 was released in 2002.It includes some complex chromosome components inherited from highly diverse parents,especially a large segment of 1RS from rye which represents a 1BL/1RS wheat-rye translocation.KN9204 was derived from the cross of five parents,including ‘Xiaoyan 693’ (XY693) (OctoploidTrititrigia,2n=56),‘Aifeng 3’ (AF3),‘Mianyang 75-18’(MY75-18),‘Xiaoyan 5’ (XY5) and ‘Jimai 38’ (JM38) (Appendix A).Of these five,Aifeng 3 is an excellent wheat variety with short plant height,lodging resistance,and high yield.Jimai 38 is a semi-winterness wheat variety,which has high resistance to stripe rust and leaf rust,and it also has the characteristics of cold resistance and lodging resistance.‘Mianyang 75-18’ is a highgeneration wheat line with excellent integrated yield characteristics.XY693 is a wheat–Th.ponticumpartial amphiploid (2n=56=40W+16E) (Lietal.2021) advanced line that possesses desirable agronomic characteristics such as disease-resistance,drought-tolerance,high yield potential,and others.XY5 is the sister line of commercial variety Xiaoyan 6 (Registration number,GS02012–1990 or GS02008–1984) derived from a distant hybridization betweenTriticumaestivumandElytrigiaelongata,which has excellent traits of disease resistance,high and stable yield potential,and good quality.To date,dozens of commercial varieties or advanced lines have been derived from KN9204 as mentioned above.
Another common wheat variety,‘Jing 411’ (J411) (Registration number: GS02002-1992),was derived from the cross between Fengkang 2 and Changfeng 1,and it has wide adaptability in the Chinese northern winter wheat region.It has a normal chromosome 1B rather than the 1BL/1RS wheat–rye translocation line.Compared to J411,KN9204 is a semi-dwarfing variety with shorter uppermost internode (UI),short but wider flag leaf,smaller seed size and a higher percentage of ear-bearing tillers (Cuietal.2014,2016,2017; Fanetal.2015,2019; Zhangetal.2017).Due to great differences in agronomic performance,including yield-related traits and the response to nitrogen deficit,a recombinant inbred line population (RIL) comprising 188 lines has been developed by crossing KN9204 with J411 through single seed descent (Cuietal.2014).This RIL mapping population was mainly used for the identification of QTLs for yield-related traits in this study,in order to specify the genetic basis of high yield potential in KN9204.
The 188 KJ-RILs together with the two parents were phenotyped for yield-related traits in 2–12 environments: 2011–2012 in Shijiazhuang,(L1); 2012–2013 in Shijiazhuang (L2),Beijing (L3) and Xinxiang (L4); 2013–2014 in Shijiazhuang (L5); and 2014–2015 in Shijiazhuang (L6).An LN treatment and an HN treatment were applied in each trial for a total of 12 environments,designated as E1 through E12,respectively (Cuietal.2014,2016,2017; Fanetal.2015,2019; Zhangetal.2017).The tiller number before winter (TNBW) was only evaluated in two different environments.A total of 22 yield-related traits were used for the QTL analysis (Appendix B).The QTL information for many of these were reported previously,such as for kernel length (KL),kernel width (KW) and thousand-kernel weight (TKW) in Cuietal.(2016); for heading date (HD),spike length (SL),spikelet number per spike (SNPS) and sterile spikelet number per spike (SSNP) in Fanetal.(2019); for flag leaf length (FLL),flag leaf width (FLW) and flag leaf area (FLA) in Fanetal.(2015); for plant height (PH),UI and the first internode length from the top (FIRITL) in Zhangetal.(2017); and for yield per plant (YPP) in Cuietal.(2014).However,all the above mentioned QTL positions in previous reports were the genetic positions rather than the physical positions.QTL information for above-ground biomass (AB),harvest index (HI),the percentage of ear-bearing tiller (PET),kernel weight per spike (KWPS),kernel number per spike (KNPS),spike number per plant (SNPP),straw dry weight (SDW) and tiller number before winter (TNBW) are first reported in this study.Herein,we report the physical position for the QTLs based on the KN9204 genome assembly for the 22 yieldrelated traits for the first time.
For AB,a plant from each plot was harvested from above the ground and then weighed.For HI,the specific value of grain weight to above-ground biomass was determined.For SDW,after threshing,all components were oven-dried at 105°C for 30 min and afterwards at 80°C for about 48 h until a constant weight was reached.For PET,the specific value of spike number to the highest stem number was determined.KNPS was measured by calculating the total number of kernels per spike of the main shoot for each plant.SNPP was evaluated by calculating the total number of spikes for each plant.TNBW was evaluated by calculating the total number of tillers for each plant,and KWPS was evaluated after harvest by weighing the main stem ears for each plant.
The phenotypic values of all 22 traits in each individual environment were used for QTL identification by IciMapping 4.1 (http://www.isbreeding.net/).The physical positions of markers in KN9204,rather than the genetic positions,were used for QTL mapping analysis,in order to facilitate the further genomic and genetic analysis of the target QTL based on the KN9204 physical map.The walking speed chosen for all QTL was 0.01 Mb,and theP-value inclusion threshold was 0.001.The threshold of LOD scores was evaluated using 1 000 permutations with a type I error of 0.05.Each significant LOD peak was counted as one individual significant QTL signal regardless of whether their physical positions overlapped or not.
2.2.High-density molecular map
A high-density genetic map with 119 566 loci spanning 4 424.4 cM has been documented (Cuietal.2017).We used the Basic Local Alignment Search Tool (BLAST) (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/release/) to align the SNP probes to the KN9204 genome.Compared to the high-density genetic map (Cuietal.2017),we reconstructed the 1B physical map and supplemented 11 154 1B-specific SNP in this study,in order to screen the QTL on chromosome 1B.
2.3.Re-sequencing of KN9204 parental lines and its derivatives
High throughput whole-genome re-sequencing (10×) was performed on KN9204,its parental lines including XY693,XY5,AF3,MY75-18 and JM38,as well as its derivatives including ‘Kenong 199’ (KN199) and ‘Kenong 2011’ (KN2011).We aligned the Illumina reads to the KN9204 genome using BWA (bwa-0.7.17) and used samtools with the setting “-q 30”,to filter out reads that did not map or were mapped in a repeat region.The alignment results were sorted and indexed by samtools (samtools-1.8).The variation calling was carried out by combining samtools mplineup,bcftools and GATK VariantFiltration.We chose SNPs with “PASS” and “SnpCluster” filter tags from the vcf output file.Based on the comparisons,we obtained the genetic composition map of KN9204 from its parental lines.Also,the origins of KN9204 elite segments could be analyzed based on the information of both QTL position and the genotypic map of KN9204.
To further trace the transmissibility of key genomic regions harboring major stable QTL,the Axiom Wheat 660K Genotyping Array was used for screening the 42 excellent advanced lines derived from KN9204,its four parental lines (XY693,XY5,AF3,and JM38),and three important main varieties: Jimai 22 (JM22,registration number: GS2006018),Liangxing 99 (LX99,registration number: GS2006016) and Shi 4185 (S4185,registration number: GS2010014).Detailed information of the 42 KN9204 derivatives is shown in Appendix C.
3.Results
3.1.Genomic coverage of the mapped SNP in the KN9204 genome
A total of 119 920 SNPs which covered 99.49% of the whole KN9204 genome (Appendix D),were consistently assigned to identical chromosomes.Of these,11 154 SNPs on chromosome 1B are reported here for the first time,169 of which were 1RS-specific SNP markers (Appendix E).To make these SNPs operational for QTL mapping analysis,only one SNP per 0.1 Mb was randomly selected from each chromosome,resulting in a total of 25 522 SNPs after eliminating the redundancy.The average distances between adjacent bin markers ranged from 0.27 Mb for 1A to 2.38 Mb for 6D,with an average of 0.56 Mb per marker pair.This high-density physical map will ensure the accuracy of QTL detection including both the QTL LOD peak position and the total QTL number.
3.2.QTL for yield-related traits identified in the KJRlL mapping population
A total of 968 QTL signals for the 22 yield-related traits were identified (Fig.1; Appendix F).The largest number of QTL signals was identified for PH,followed by TKW and SL; and the lowest number of QTL signals was identified for TNBW (Appendix G).TNBW was only evaluated in two different environments,thus resulting in the lowest number of QTL signals.The 968 QTL signals harbored 31 stable QTLs for 14 yield-related traits,which showed a similar LOD peak position (less than 10 Mb) in no less than four different environments.These stable QTLs were distributed on chromosomes 1B,2A,2B,2D,3A,3D,4A,4B,5A,5B,5D,6B,7A,and 7D (Appendix H).KN9204 contributed alleles for 15 (48.4%) of the 31 stable QTLs.Considering the overlap of confidence intervals across environments,the 968 QTL signals were grouped into 589 individual QTLs.Of them,169 (28.7%) QTLs were reproducibly identified in no less than two environments.KN9204 and J411 contributed 71 (42.0%) and 98 (58.0%) favorable alleles of the 169 environment-repeatable QTLs (Appendices F and I).Favorable QTL alleles from KN9204 and J411 were distributed randomly on the wheat genome in most cases.In addition,many co-locating QTLs were detected,which are listed in Appendix I.
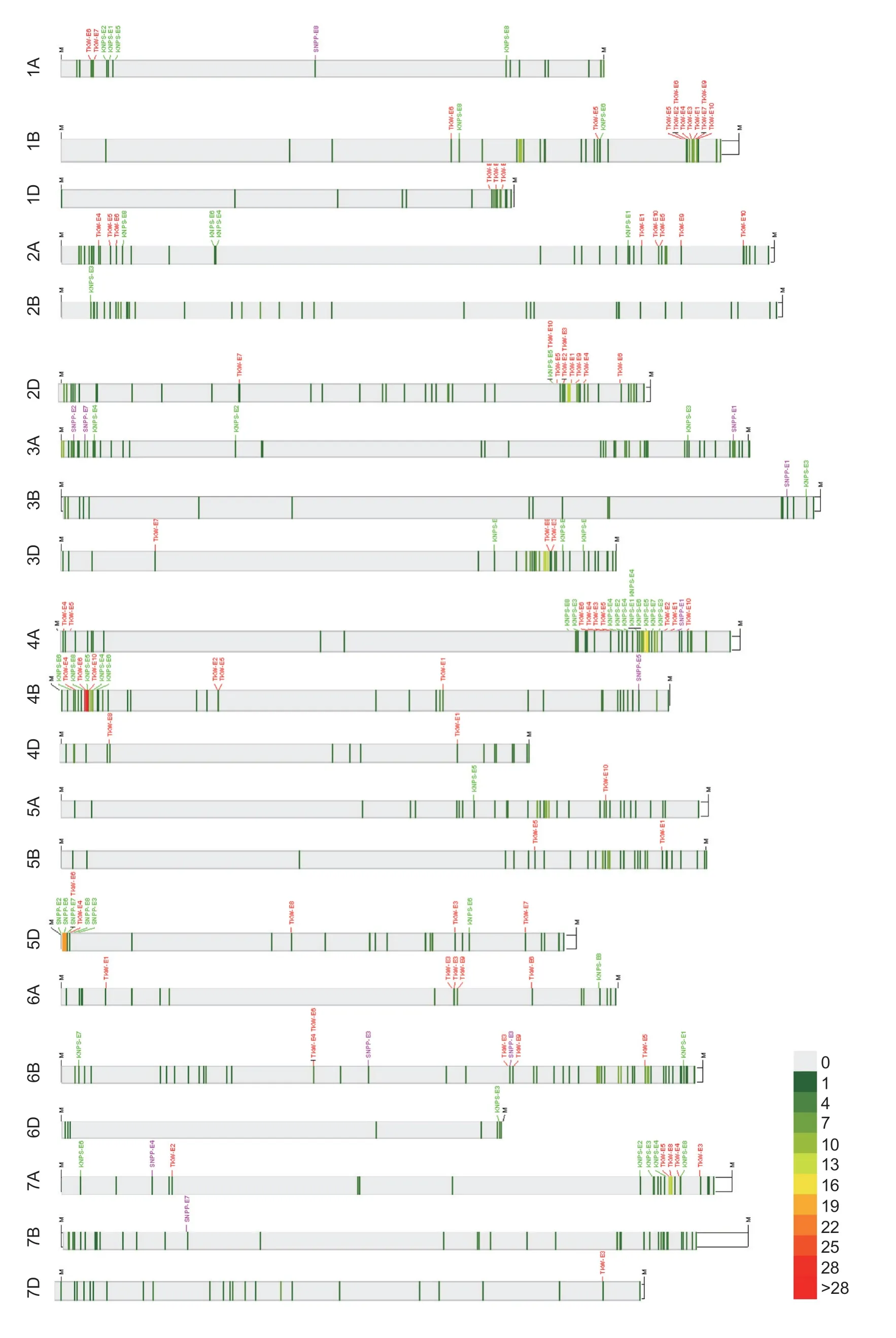
Fig.1 Distribution of QTL signals for the 22 yield-related traits on the 21 wheat chromosomes.The number of QTL signals per 5 Mb in the corresponding chromosomal regions (LOD peak position) are shown in different colors.Three elements of yield (thousand-kernel weight (TKW),kernel number per spike (KNPS) and spike number per plant (SNPP) are listed and shown in the different colors.
In general,J411 contributed more favorable QTL signals than KN9204,55.0%vs.45.0% (Appendix G).Among the 22 yield-related traits,KN9204 contributed to more excellent QTL signals for YPP,HI,FLL,SL,PET,and SNPS; while J411 contributed to more favorable QTL signals for FIRITL,UI,SNPP,KW,KWPS,SDW,KNPS,KL,TNBW,TKW and SSNP.
3.3.Genetic composition of KN9204 based on the re-sequencing results
Based on the high throughput whole-genome resequencing,we obtained the genotypic map of KN9204 from its parental lines (Fig.2).A total of 3 313 recombination events were identified.Large numbers of recombinant events occurred on chromosomes 1B,2A,3B,4A,5A,7B and 7D,whereas fewer occurred on chromosomes 1A,2B,6A and 6B (Appendix J).
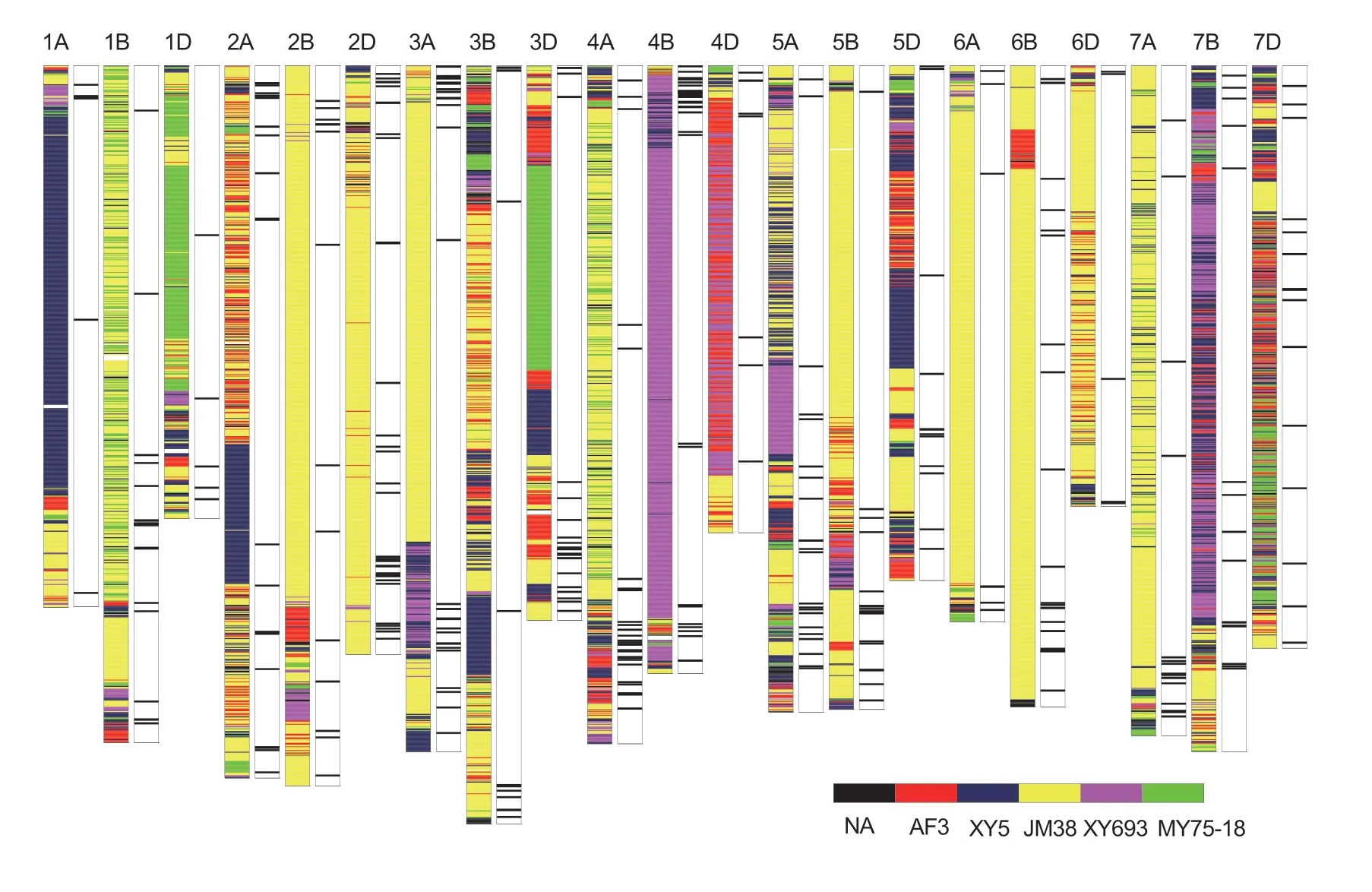
Fig.2 Genotypic map of ‘Kenong 9204’ (KN9204) and the distribution of favorable QTLs from KN9204 on the 21 wheat chromosomes.For each chromosome,the colored histogram on the left indicates the genotypic map of KN9204.Chromosomal regions with different colors indicate the regions inherited from each of its parents.NA,missing data; AF3,Aifeng 3; XY5,Xiaoyan 5; JM38,Jimai 38; XY693,Xiaoyan 693; MY75-18,Mianyang 75-18.The black–white histogram on the right indicates a QTL LOD peak region (2 Mb flanking the LOD peak position) with the favorite additive effects from KN9204.
Overall,50.33,14.82,11.31,10.82,and 9.41% of the genomic regions in KN9204 were inherited from JM38,XY5,AF3,XY693,and MY75-18,respectively (Appendix K).These results are generally in accordance with its pedigree tree (Appendix A).We compared the genetic contribution ratios from each parental line to KN9204 between the expected and observed values (Fig.3-A).The results showed that the proportions of alleles inherited from AF3,XY693 and XY5 were 80.96,73.12 and 18.56% higher than the expected values.AF3,XY693 and XY5 contributed 11.93,16.97 and 13.99% of the favorable QTL alleles in KN9204,respectively (Appendix L).XY693 is a wheat–Th.ponticumpartial amphiploid advanced line that has excellent agronomic characteristics.Approximately 38.0,17.7 and 13.2% of the genomic regions from XY693 in KN9204 were distributed on chromosomes 4B,7B and 4D,respectively (Fig.3-B).The QTL information showed that XY693 contributed 23.9% of the 71 environmentrepeatable QTL with favorable alleles from KN9204 (Appendix F).Taking chromosome 4B as an example,39 (66.1%) of the 59 QTL signals with positive effects from KN9204 were contributed by XY693,including four major stable QTLs:qFlw-4B(E1/E2/E4/E5/E6/E7/E8),qKnps-4B(E4/E5/E6/E8),qPh-4B(E1/E2/E3/E4/E5/E6/E7/E8/E9/E10),andqSsnp-4B(E2/E5/E6/E7/E8) (Fig.3-C and D;Appendix F).In addition,a major stable QTL for the maximum root length (MRL),qMrl-7B,had been identified on chromosome 7B (Fanetal.2018).The genotypic map of KN9204 showed that XY693 contributed to the favorable alleles ofqMrl-7B(Fig.3-E).The above results indicated that KN9204 is a variety that has integrated numerous key genomic regions from each of its parental lines,especially from the wheat–Th.ponticumpartial amphiploid XY693.
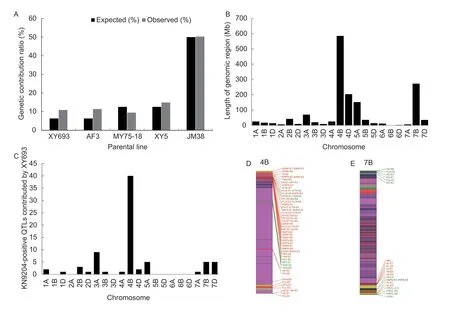
Fig.3 Distribution of the genomic regions from ‘Xiaoyan 693’ (XY693) to ‘Kenong 9204’ (KN9204) and their effects on yield-related traits.A,comparison of the genetic contribution ratios from the parental lines to KN9204 between the expected values and the observed values.The expected proportions of alleles were calculated assuming that alleles conform to Mendelian inheritance,with a 50:50 transmission probability of alleles from parent to offspring.B,distribution of the genomic regions from XY693 to KN9204.C,genomic distribution of excellent QTL signals from XY693 to KN9204.D,genotypic map of chromosome 4B of KN9204 and the distribution of the environment-repeatable QTLs for yield-related traits.Favorable QTL alleles from KN9204 and J411 are shown in red and green,respectively.E,genotypic map of chromosome 7B of KN9204 and the distribution of the environment-repeatable QTL for yield-related traits.Favorable QTL alleles from KN9204 and J411 are shown in red and green,respectively.
3.4.Fine mapping of major stable QTL from the wheat–Th.ponticum partial amphiploid XY693
To characterize the genetic effects of key genomic regions from XY693,we performed fine mapping and genetic analysis of the major stable QTLs on chromosomes 4B and 7B,which benefited from the high-resolution molecular map and the assembled genome of KN9204.
For plant height (PH),J411 was significantly higher than KN9204 in all 10 of the investigated environments (Zhangetal.2017).A major stable QTL for PH (qPh-4B)was identified on chromosome 4BS in all 10 environments,with LOD peaks in 29.10–31.60 Mb in the KN9204 physical map (Fig.4-A; Appendix H).Moreover,two major stable QTLs for FLW (qFlw-4B) and KNPS (qKnps-4B) were also identified in this genomic region (Appendix H).

Fig.4 Fine mapping of qPh-4B and its candidate gene predictions.A,LOD profiles of qPh-4B in 10 investigated environments,where the blue dotted line indicates the position of the known green revolution gene Rht-B1,and the colored rectangle indicates the genotypic map of ‘Kenong 9204’ (KN9204) in the corresponding segment,chromosomal regions with different colors indicate the regions inherited from each of its parents,black represents missing data,red represents ‘Aifeng 3’,blue represents ‘Xiaoyan 5’,yellow represents ‘Jimai 38’,purple represents ‘Xiaoyan 693’,and green represents ‘Mianyang 75-18’.B,spatiotemporal expression patterns of TraesKN4B01HG04430 in KN9204 and J411.C,fine mapping of qPh-4B using the 188 KJ-RILs as secondary mapping populations.The plant heights (PH) in the 188 KJ-RILs were evaluated in 10 environments,and five of each were investigated under low nitrogen (LN) and high nitrogen (HN) conditions,respectively.The averaged PH of each line under the LN and HN conditions were used for fine mapping analysis of qPh-4B.For each histogram,the left indicates the average PH under LN and the right indicates that under HN.The colorful rectangles below the histogram indicate the recombinants at the target region,and alleles from KN9204 and J411 are marked in red and green,respectively.Below the colorful rectangles are the numbers of the corresponding recombinants.The letters at the bottom of the figure indicate the genotypes of qPh-3D,qPh-5A and qPh-6B; and AA indicates that the alleles were identical to KN9204,while those marked BB were identical to J411.Bars mean SD (n=25).D,open reading frame sequence differences of TraesKN4B01HG04430 between KN9204 and J411.
The well-known green revolution gene Rht-B1 encoding DELLA proteins is located on chromosome 4BS (Peng et al.1999).We aligned Rht-B1 to KN4B: 31 395 925–31 397 423 in the KN9204 assembly,and it overlaps with the LOD peak of qPh-4B (Fig.4-A),so Rht-B1 might be the candidate gene of qPh-4B.To verify this prediction,fine mapping ofqPh-4Bwas performed using the 188 KJRILs as secondary mapping populations.A total of four major stable QTLs,i.e.,qPh-3D,qPh-4B,qPh-5AandqPh-6B,led to the differences of PH between KN9204 and J411 (Appendix H).We divided the 188 KJ-RILs into eight groups based on the genotypes ofqPh-3D,qPh-5AandqPh-6B(Fig.4-C).The KJ-RILs within each group could be regarded as QTL near isogenic lines (QNILs) ofqPh-4B.The eight sets of QNILs narrowedqPh-4Bto a 0.69-Mb physical region (KN4B: 31.15–31.84) between markersAX-9514567andAX109284839,which contains nine predicted high-confidence genes in KN9204 (data not shown).Four differentially expressed genes (DEGs) existed in this region based on the transcriptomic data of KN9204 and J411 (Fig.4-B; Appendix M).Of these,TraesKN4B01HG04430(KN4B: 31 395 636–31 397 864) was expressed at significantly different levels in leaf (heading stage),stem (booting stage),and spike (jointing stage and heading stage) under normal N conditions,and in leaf (heading stage),spike (heading stage) and seed at 21 days under low N stress (Fig.4-B),and it is predicted to encode a DELLA protein corresponding toTraesCS4B02G043100.A comparison of the KN9204 genomic sequences and J411 (Pseudomolecules of J411 based on 40× resequencing datasets) showed that a T to C substitution (T in KN9204vs.C in J411) resulted in the allelic variation ofRht-B1between KN9204 (Rht-B1b) and J411 (Rht-B1a) (Pengetal.1999) in the open reading frame (ORF) ofTraesKN4B01HG04430(Fig.4-D).The three remaining DEGs had no sequence differences in the ORFs.All the above results confirmed thatTraesKN4B01HG04430is the causal gene ofqPh-4B,i.e.,Rht-B1.
Similar toqPh-4B,qFlw-4BandqKnps-4Bwere fine mapped to KN4B: 28.68–30.01 Mb and KN4B: 32.73–38.93 Mb in KN9204,respectively (Appendices N and O).Both regions excluded the gene ofRht-B1,indicating thatqFlw-4BandqKnps-4Bmight be in close linkage but not the pleiotropy ofRht-B1.
3.5.Transmissibility analysis of 12 favorable stable QTLs from KN9204 to its derivatives
The 119 920 polymorphic SNPs between KN9204 and J411 were used to perform a genetic diversity analysis of KN9204,its four original parents (excluding MY75-18 due to the missing genotype information),its 42 derivatives,J411 and three Chinese main varieties (Appendix P).The results showed that the genetic similarity coefficients among the 51 varieties/advanced lines ranged from 0.40 to 1.00.Some genetic differences existed among the 42 KN9204 derivatives,except for the five groups of the advanced sister-lines.
A total of 16 stable QTLs,includingqMrl-7B(maximum root length,which was first mentioned in Section 3.3) with favorable alleles from KN9204,were identified on 12 genomic segments that were labeled segment 1 (S1) to segment 12 (S12) (Appendix H; Fig.5).The genomic segments covering all LOD peaks across the environments were regarded as the key genomic segments,which represented the corresponding favorable QTL by breeders and were used for further analysis.Based on the SNP genotypic scores,genotypic maps of the 12 key genomic segments in J411,KN9204,its original parents,its 42 derivatives and three Chinese main varieties were constructed (Fig.5).
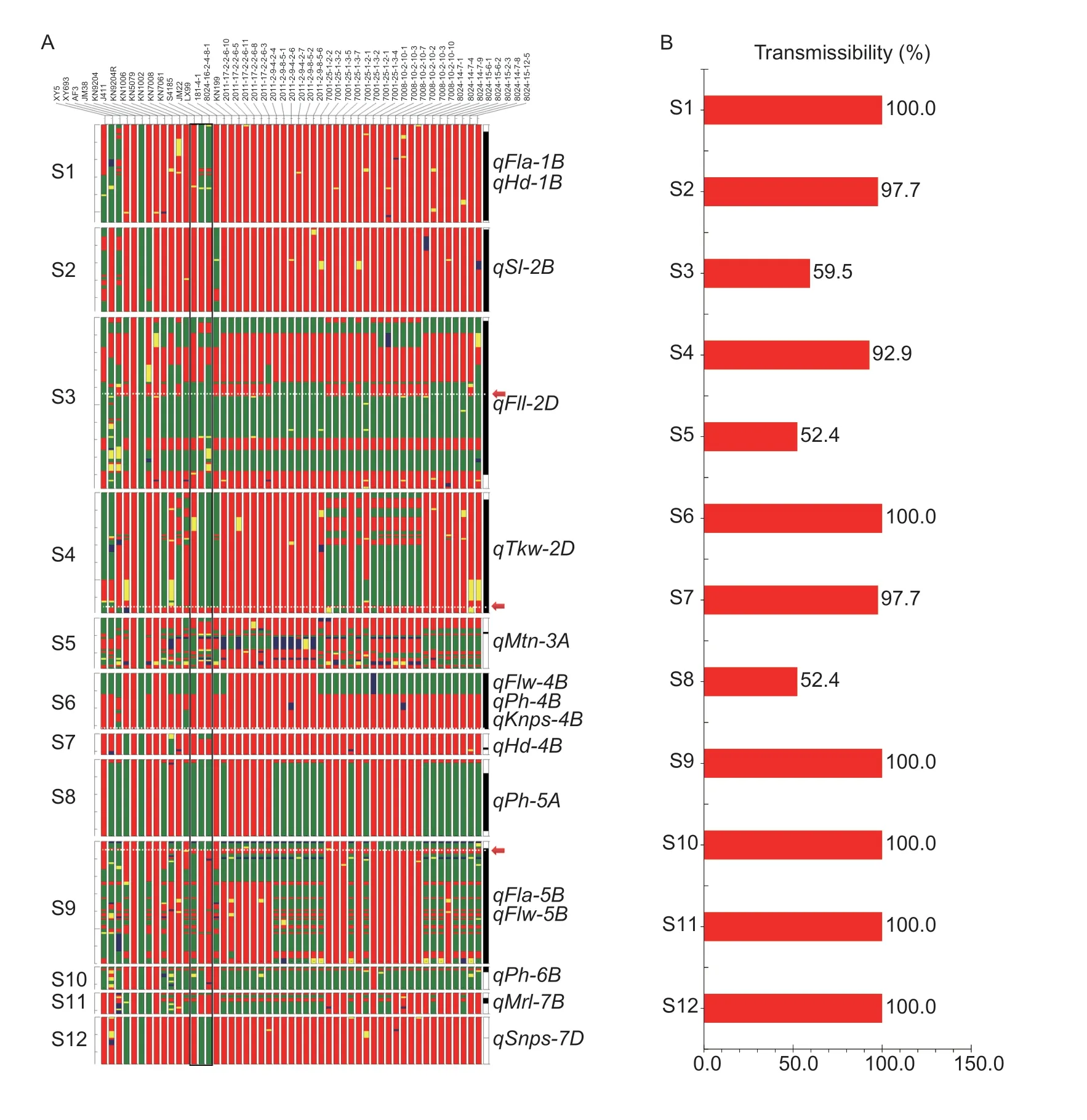
Fig.5 Twelve key genomic segments regions (S1–S12) harboring stable QTLs with favorable alleles from ‘Kenong 9204’ (KN9204).A,genotypic map of J411,KN9204,its original parents,its 42 derivatives and three Chinese main varieties in the 12 key genomic regions.B,transmissibility analysis of the 12 key genomic segments.The colored histograms represent the 12 key genomic regions (S1 to S12) harboring stable QTL.Genomic regions in red and green indicate that the corresponding variety/advanced lines in the corresponding regions possessed alleles identical to KN9204 and J411,respectively.Genomic regions with heterozygous genotypes and missing values are shown in blue and yellow,respectively.The right-most black-and-white histograms in black indicate the most likely positions of the corresponding QTL based on the QTL LOD peak position.For S3,S4,and S9,the arrows in red along with the dotted lines in white indicate the most probable positions of the corresponding QTL.The three histograms enclosed with black boxes indicate the genotypic map of the three Chinese main varieties,i.e.,Shi 4185,Jimai 22,and Liangxing 99.
For segments S1,S2,S7 and S12,the whole key genomic segments were passed onto all 42 of the KN9204 derivatives,with the exceptions of S2 in KN9204R and S7 in KN1002.For S10,the LOD peak positions ofqPh-6Branged from 646.65 to 649.15 Mb,and it was mapped to 649.05 Mb in six of eight (75.0%) environments; and the segments of 648.88–649.018 Mb from KN9204 were passed onto all 42 of its derivatives.For both S5 and S8,22 of the 42 (52.4%) derivatives inherited the key genomic segments from KN9204.For S3,the LOD peak positions ofqFll-2Dwere mapped to 14.35,18.35,18.45,and 23.15 Mb in four different environments; thus,the candidate genes ofqFll-2Dwere most likely in the segments of 18.14–18.56 Mb,and this segment from KN9204 was passed onto 25 of the 42 (59.5%) KN9204 derivatives.For S4,the LOD peak positions ofqTkw-2Dwere mapped to 548.46,549.46,554.96,and 554.96 Mb in four different environments; thus,the candidate genes ofqTkw-2Dwere most likely in the segments of 554.68–555.00 Mb,and this segment from KN9204 was passed onto 39 of the 42 (92.9%) KN9204 derivatives.For S6,qPh-4Bshould have the effects ofRht-b1that were located in 31.395925–31.397423 Mb in the KN9204 assembly,and this segment from KN9204 was passed onto all 42 of the KN9204 derivatives.In addition,qFlw-4BandqKnps-4Bwere fine mapped to KN4B: 28.68–30.01 Mb and KN4B: 32.73–38.93 Mb in KN9204 (Appendices N and O),and both segments from KN9204 were passed onto all 42 of the KN9204 derivatives.For S9,the LOD peak positions ofqFla-5Bwere mapped to 599.42,599.42,602.32,605.22,and 606.92 Mb in five different environments;qFlw-5Bwas mapped to 599.42,599.42,599.42,605.22,and 607.22 Mb in five different environments; and this segment from KN9204 was passed onto all 42 of the KN9204 derivatives.qMrl-7Bwas narrowed down to the region of KN7B: 597.2–597.4 Mb(data not shown),and this segment from KN9204 was passed onto all 42 of the KN9204 derivatives.Taken together,the vast majority of the segments with favorable alleles from KN9204 were preferentially selected in the process of breeding selection (Fig.5).
For the three Chinese main varieties,nine (S1,S2,S4,S6,S7,S9,S10,S11,and S12),eight (S2,S4,S5,S6,S7,S9,S10,and S11),and eight (S2,S4,S5,S6,S7,S9,S10,and S11) key genomic segments of S4185,JM22,and LX99 were identical to KN9204 (Fig.5).These results indicated that most of the 12 key genomic segments have experienced strong selection pressures and have been effectively fixed in wheat breeding programs.
4.Discussion
4.1.Candidate foundation parents inherited genomic regions from their parental lines selectively
Some have suggested that,to a large extent,progress in wheat breeding has been a result of the conservation of favorable linkage blocks as well as the assemblage of beneficial linked epistatic gene interactions (Holland 2001; Hanetal.2020).Candidate foundation parents generally have more excellent characteristics such as high yield potential,ideotype,excellent processing quality,good disease resistance,and others.Therefore,much more directionally intensive artificial selection has been experienced in the process of breeding candidate foundation parents.Generally,inheritance is random in the absence of selection in the process of crossbreeding,so each parent contributes to the genetic materials of its derivatives equally and the outcome conforms to the genetic laws of Mendel (Hanetal.2020).However,intensive artificial selection results in significant deviations from the predictions of classical Mendelian inheritance theory in the parental genetic contributions to elite varieties,especially for candidate foundation parents (Fradgleyetal.2019; Hanetal.2020; Haoetal.2020; Renetal.2021).
The proportions of alleles inherited from AF3,XY693 and XY5 were higher than the expected values.This indicated that genomic regions from AF3,XY693 and XY5 might have undergone strong selection pressure in the breeding process since they harbor numerous excellent alleles.XY693 contributed 16.97% of the favorable QTL alleles in KN9204 (Fig.3),which is significantly (171.52%) higher than expected.Therefore,XY693 should have played key roles in KN9204 as a candidate foundation parent.Recently,Lietal.(2021) confirmed that XY693 contains 16Th.ponticumchromosomes and 40 wheat chromosomes,with the absence of a chromosome 6B pair.Approximately 70.0% of XY693’s genomic segments in KN9204 are distributed on chromosomes 4B,7B and 4D (Fig.3),highlighting the importance of these genomic segments which probably harbor excellent important genes or favorable allelic variations.For chromosome 4B,66.1% of the 59 favored QTLs from KN9204 were contributed by XY693,including the four major stable QTLs ofqFlw-4B,qKnps-4B,qPh-4B,andqSsnp-4B.XY693 made no genetic contributions to KN9204 on chromosome 6B,which is consistent with the absence of a 6B chromosome pair in XY693 (Lietal.2021).
The genetic contribution ratio of MY75-18 to KN9204 was 24.72%,which is lower than that of the expected value.This may be attributed to the diversity of ecological types between MY75-18 and KN9204.MY75-18 was bred in southwest China and thus harbors limited excellent haplotypes that would be adaptive for growth in the Huang–Huai Winter Wheat Area in China.The observed proportion of alleles inherited from JM38 in KN9204 was as expected,conforming to the genetic laws of Mendel.The above results confirmed that the candidate foundation parents inherited genetic materials from their parental lines selectively.
KN9204 and J411 are important candidate foundation parents in China,both of which should harbor numerous excellent genes or favorable allelic variations.QTL analysis based on KJ-RILs showed that KN9204 and J411 contributed equally to the favorable QTL alleles (Appendices F and I).This is consistent with their excellent agronomic performance,in which KN9204 performed better in yield potential such as harvest index,spikes per unit,plant height and leaf erectness,while J411 performed better in thousand-kernel weight,kernel width,kernel length,kernel weight per spike,spike number per plant,and other characteristics.Intriguingly,favorable QTL alleles from KN9204 did not co-localize with those from J411 in most cases,excluding the possibility of adverse genetic linkage or linkage drag of the target QTL (Appendix I).Both varieties have undergone strong selection pressures in the process of their breeding programs,which might account for the breakup of the adverse genetic linkage.
4.2.Candidate foundation parents harbor traceable genomic regions with hitchhiking effects in their derived varieties
The diversity of loci under strong positive selection pressure is significantly lower than that of other loci in a natural population,as well as in a particular breeding population.Diversity in the genomic regions flanking these specific loci also declines in the process of selection,which is called “hitchhiking effects” or “selection sweep” in genetics (Andolfattoetal.2001; Schl?ttereretal.2003).Directionally intensive artificial selection by breeders results in the conservation of excellent haplotype blocks in crop breeding programs.Therefore,the selection sweeps are regarded as diagnostic for genomic regions that contain genes controlling important economic traits that are selected by domestication and breeding (Hanetal.2020; Haoetal.2020; Renetal.2021).Key genomic regions harboring excellent genes/QTLs from candidate foundation parents tend to accumulate in the most recently established varieties in breeding programs under intensive artificial selection by breeders.
KN9204 is a candidate foundation parent that has dozens of new derived varieties or advanced lines.Thus,KN9204 genomic regions that have been stably transmitted to its derivatives should be essential for the excellent performance of KN9204 and its derivatives.Nine (75.0%) of the 12 genomic regions harboring key excellent QTLs from KN9204 could be stably transmitted to its derivatives,with transmissibility up to 90.0% (Fig.5).This suggests that breeder selection benefits the conservation of favorable haplotypes and linkage blocks.The potential values of these nine chromosomal segments in molecular breeding programs should be great.
We found that some small genomic regions were easier to selectively fix than the large ones in the derived varieties,which was evidenced by S9 and S10 (Fig.5).For S9,many recombinant events occurred in this genomic region (approximately 6.95 Mb)in the KN9204 derived varieties.However,a small genomic segment (approximately 0.12 Mb) closely linked withqFla-5BandqFlw-5B,two co-located major stable QTLs for increasing flag leaf area and width,was preferably selected and fixed in all 42 of its derived varieties.Though 89.3% of the 1.31 Mb genomic region in S10 was identical to J411 rather than KN9204 in all 42 of its derivatives,a small genomic region (approximately 0.14 Mb) closely linked withqPh-6B,a major stable QTL for reducing plant height,was preferably selected and fixed in all its derived varieties.The above results confirmed that it is possible to trace the key genomic regions,even relatively small segments,in a candidate foundation parent with the advanced high throughput molecular marker technology.
In addition,genomic regions harboring adverse QTL alleles in KN9204 were found to be replaced by those of J411,i.e.,the favorable alleles,in the 42 KN9204-derived lines/varieties in most cases.For example,qTkw-1Bwas mapped to KN1B: 692.4–700.3 Mb in eight different environments (Appendix F).We have narrowedqTkw-1Bto a physical distance of 2.3 Mb based on QNILs (data not shown).Intriguingly,a strong selection signal from J411 was detected in this genomic region in the 49 tested advanced lines/varieties (Fig.6).These results indicated that favorable alleles from J411 have been selected and fixed gradually in wheat breeding programs.
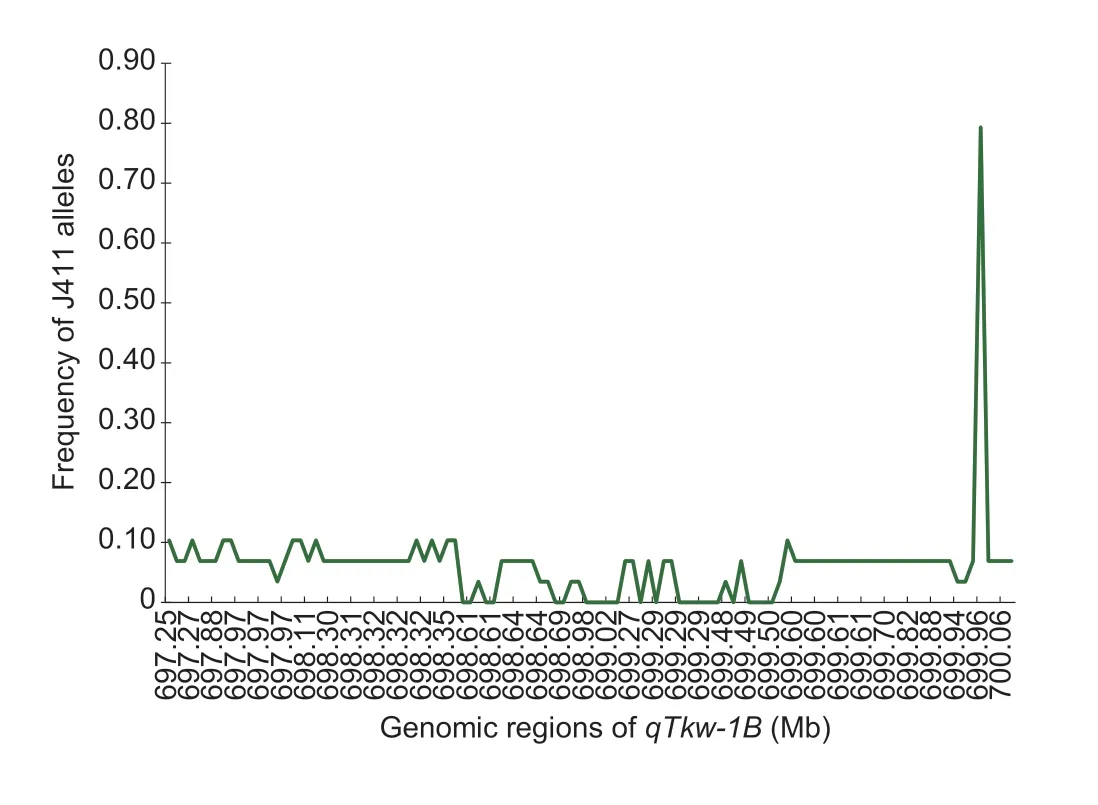
Fig.6 Frequency of J411 alleles in the target region of qTkw-1B (approximately 2.3 Mb interval) in 42 excellent advanced lines derived from Kenong 9204 (KN9204).
4.3.QTL identification and isolation will facilitate 5G breeding programs
Phenotype-based selection in traditional breeding programs has resulted in the great phenotypic changes eventually seen in modern crops (Zhuang 2003; Doebleyetal.2006; Meyeretal.2012).However,lower selection accuracy along with limited genetic gain in conventional breeding programs cannot meet the increasing demand for food for the increasing population of approximately 9 billion people by 2050,especially for complex traits under the control of quantitative trait loci (Zhuang 2003; Godfrayetal.2010; Rayetal.2013; FAO 2019).The 5G breeding approach might bring much needed disruptive changes to crop improvement (Varshneyetal.2019).The selection sweeps identified in candidate foundation parents could be regarded as targets for further functional genetic analysis of a genomic region containing excellent genes for economic traits,which is of great value for both map-based gene cloning and molecular breeding by a design approach.Therefore,dissecting the genetic basis of candidate foundation parents not only provides an effective approach to GFI but also provides novel gene resource for GB.We identified nine key genomic regions with strong selection traces in the derivatives of KN9204 in this study (Fig.5).Further study of the fine mapping and map-based cloning of these genes will effectively promote their utilization in 5G breeding programs.
We preliminarily performed fine mapping analysis of a QTL cluster on S6,and dissected it into three individual QTL based on the multi-omic information of the primary mapping population.To date,the secondary mapping population,such as back-crossing-derived populations or residual heterozygote-derived populations,has been widely used for fine mapping and map-based cloning of the target QTLs,but they are laborious,expensive and timeconsuming (Salvi and Tuberosa 2005).The results of this study indicated that the ultrahigh-throughput genotype screening technology along with multiple-omics platforms provide a new paradigm for identifying candidate genes and clues for the functions of their target QTLs.
The detectability of putative QTL in a bi-parental mapping population is known to be determined by the genetic differences of the two parents,to a large extent,in addition to the population size and marker density.Therefore,limited QTL could be identified in bi-parental mapping populations (Mylesetal.2009; Gaireetal.2020).In addition,QTL analysis based on a bi-parental mapping population cannot capture all the allelic diversity in germplasm (Gaireetal.2020).A natural mapping population based on genome wide association studies (GWAS) should be used to comprehensively understand the genetic basis of high yield potential in KN9204 in the future.
5.Conclusion
We released the first high-resolution genetic composition map of the candidate foundation parent KN9204.Major stable QTLs for yield-related traits were identified,and the transmissibility of key chromosomal segments with positive effects from KN9204 were specified.Evidence for selection sweeps in KN9204 and its derivatives was verified.The results of this study will facilitate 5G breeding programs for the genetic improvement of yield potential in wheat.
Acknowledgements
This research was jointly supported by the grants from the Shandong Major Basic Research Project of Natural Science Foundation,China (ZR2019ZD16),the Shandong Provincial Key Research and Development Program,China (2019GNC106126 and 2021LZGC009),the Natural Science Foundation of Hebei Province,China (C2021205013),the Hebei Scientific and Technological Innovation Team of Modern Wheat Seed Industry,China (21326318D),the National Natural Science Foundation of China (31871612,31901535,and 32101726),and the China Agriculture Research System of MOF and MARA (CARS-03).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on https://doi.org/10.1016/j.jia.2023.02.013
 Journal of Integrative Agriculture2023年9期
Journal of Integrative Agriculture2023年9期
- Journal of Integrative Agriculture的其它文章
- Combining nitrogen effects and metabolomics to reveal the response mechanisms to nitrogen stress and the potential for nitrogen reduction in maize
- Natural variations and geographical distributions of seed carotenoids and chlorophylls in 1 167 Chinese soybean accessions
- Carbon sequestration rate,nitrogen use efficiency and rice yield responses to long-term substitution of chemical fertilizer by organic manure in a rice–rice cropping system
- ldentification and epitope mapping of anti-p72 single-chain antibody against African swine fever virus based on phage display antibody library
- Dissecting the genetic basis of grain color and pre-harvest sprouting resistance in common wheat by association analysis
- The PcERF5 promotes anthocyanin biosynthesis in red-fleshed pear (Pyrus communis) through both activating and interacting with PcMYB transcription factors