
Genome characterization of the Caprine arthritis-encephalitis virus in China: A retrospective genomic analysis of the earliest Chinese isolates
2023-03-11 06:46:56WANGDengfengYANGXueyunWEIYurongLIJianjunBOLATIHongduziMENGXiaoxiaoTUERXUNGunuerNUERDANNuerbaihetiWUJianyong
WANG Deng-feng ,YANG Xue-yun ,WEI Yu-rong ,LI Jian-jun ,BOLATI Hongduzi ,MENG Xiao-xiao,TUERXUN Gunuer,NUERDAN Nuerbaiheti,WU Jian-yong#
1 Engineering Laboratory of Animal Pharmaceuticals,College of Animal Science (College of Bee Science),Fujian Agriculture and Forestry University,Fuzhou 350002,P.R.China
2 Xinjiang Key Laboratory of Animal Infectious Diseases,Institute of Veterinary Medicine,Xinjiang Academy of Animal Sciences,Urumqi 830013,P.R.China
Abstract Caprine arthritis-encephalitis virus (CAEV) is an under-studied virus infecting caprines and ovines worldwide. Over the last four decades,CAEV has spread in China,obtaining genomic data on CAEV strains circulating in China is of importance for developing diagnostic methods and eradicating associated diseases. However,there is limited information on the genome,including characterizations,and the probable origin. This work aimed to characterize Chinese CAEV genomes and population structures. Five CAEV strains isolated from infected dairy goats between 1989 and 1994 in Gansu,Guizhou,Shaanxi,Shandong and Sichuan provinces were cloned and sequenced. The Chinese CAEV had a 58-93% genome similarities to strains outside of China,and they belonged to subgenotype B1. The highest similarity levels (98.3-99.3%) were with two other Chinese strains,and they shared a 91.8-92.3% similarity with the strain Clements (GenBank accession no.NC_001463.1) from outside of China. The Chinese CAEV strains isolated from different provinces over five years were still highly homologous and contained unique ancestral population components,indicating that these Chinese strains had a common origin that differed from other known strains. Our results provide genomic data on circulating Chinese CAEV strains and will be useful for future epidemiological investigations and CAEV eradication programs.
Keywords: Caprine arthritis-encephalitis virus,genotype,phylogenetic analysis,population structure,similarity,dairy goat
1.Introduction
Caprine arthritis-encephalitis virus (CAEV) infections mainly cause continuous and progressive pathological damage to the joints,mammary glands,and central nervous system of goats and sheep (Pinczowskiet al.2017;Michielset al.2018). Caprine arthritis-encephalitis is listed as a Class III animal disease by the Ministry of Agriculture and Rural Affairs of the P.R.China,and the World Organization for Animal Health must be notified of the presence of this terrestrial animal disease. CAEV belongs to genusLentivirus,familyRetroviridae,and it is collectively referred to as a small ruminant lentivirus(SRLV) together with the Maedi-visna virus (MVV)(Highland 2017). Most infected goats exhibit latent infections,and only 30% of infected goats show clinical symptoms after several years of infection (Michielset al.2018). CAEV can infect goats of any age and breed,with dairy goats being the most susceptible (Gumusova and Memis 2016). The infections mostly occur during the goat lamb period. It is transmitted from female goats to lambs through lactation,or horizontally through longterm contact (Kabaet al.2012;Kurhaluket al.2021). It can cause serious economic losses to the goat breeding industry,and it is recognized as an important disease in entry-exit trade inspections and quarantine restrictions.
CAEV possess a complex genome includes three structural genes,that are,env,gagandpol. Additional reading frames encode proteins with regulatory functions in viral replication,such as vpr,rev and vif (Villetet al.2003;Bartáket al.2018). Furthermore,at both ends of the genome,long terminal repeats contain U3,R and U5 regions. The U3 region has the promoter and enhancer regions of the virus that are responsible for tissue tropism (Oskarssonet al.2007). In all the above proteins,gag protein elicits humoral immune responses that are easy to detect;therefore,antibodies against gag protein are used as diagnostic biomarkers for the routine surveillance and epidemiological investigations of Caprine arthritis-encephalitis (Picottoet al.2021). In addition to serosurveys,genotyping can be used to reveal etiological characteristics. Currently,five genotypes (A,B,C,D and E)have been determined using phylogenetic analyses based on partialgaggenes,gag-polgenes or long terminal repeat sequences,and genotypes A,B and E strains have been further divided into A1 to A22,B1 to B5 and E1 to E2 subtypes,respectively (Michielset al.2020;Molaeeet al.2020). Genotypes A and B are considered MVV-like and CAEV-like strains,respectively,and they are predominant worldwide. Group C strains have been identified in Norwegian sheep and goats,genotype D strains have been detected in sheep and goats in Switzerland and Spain,and genotype E strains have been mainly found in Italy (Shahet al.2004;Gjersetet al.2009;Reinaet al.2010).
CAEV was first isolated from goat joint synovial fluid and cornea by Crawfordet al.(1980) in the USA in 1980. CAEV infections are distributed in goats and wild ruminants worldwide (Erhoumaet al.2008;Gomez-Luciaet al.2018;Olechet al.2020). In China,CAEV was first isolated from imported Saanen dairy goats in Gansu Province,northwestern China in 1988,and then,CAEV was successively isolated from Saanen dairy goats and Toggenburg goats in Shandong,Sichuan,Shaanxi and Guizhou provinces (Baihuojiaet al.1989;Huet al.1989;Liet al.1990;Linet al.1990;Wanget al.1994). Now it is an endemic disease in China that had an individual seroprevalence of 3.33% during 1987-1995 (Gonget al.1996). A recent serosurvey showed that 0.4% of goats(excluding dairy goats) in 11 provinces of China were seropositive (Sunet al.2018). However,there are still limited sequenced CAEV genomes that were isolated from China. In this study,we aimed to characterize the five CAEV strains isolated in 1989-1994. After a genotype analysis,the epidemic genotypes of the Chinese CAEV strains were determined,and the genetic relationships and population structures of the whole genomes were analyzed.The research results provide reference genome data for future source-tracing and the control of CAEV in China.
2.Materials and methods
2.1.Viral strain
The five CAEV strains,239,CGu-3,CSI-4,gs-35V and SH-1,were isolated in China from Shandong,Guizhou,Sichuan,Gansu and Shaanxi provinces,respectively,during 1989-1994 (Table 1) (Baihuojiaet al.1989;Huet al.1989;Liet al.1990;Linet al.1990;Wanget al.1994). All five strains were stored in liquid-nitrogen.Proviral DNA was extracted from 1 000 μL of cell pellets using a QIAamp DNA Mini Kit (Hilden,QIAGEN) in accordance with the manufacturer’s instructions. Purified DNA was eluted with 200 μL of Tris-EDTA buffer.
2.2.Cloning and sequencing of the full-length viral genome
For the amplification and sequencing of whole genomes,seven primer sets (Table 2) were designed with overlapping regions based on the CAEV sequences (GenBank accession nos.AY900630.1,M14149.1 and M33677.1)using Oligo 7 Software. Purified proviral genomic DNA was subjected to PCR assays for whole genome amplification.Each 50-μL reaction contained 2 μL of DNA template,1 μL of each primer (20 mmol L-1) and 25 μL 2× PCR reaction mix (Promega,Madison,USA). Amplified fragments were visualized on 1.5% (w/v) agarose gels in TAE (0.04 mol L-1Tris-acetate and 0.001 mol L-1EDTA) buffer and purified using a Tiangen DNA Gel Extraction Kit (Tiangen,Beijing,China). The purified products were then ligated individually into the pGEM-T Easy vector (Promega,Madison,USA)at 16°C for 2 h. Ligated products were transformed into competentEscherichia coliDH5α cells (Beijing,Tiangen),and recombinant positive clones were screened on LB/ampicillin/IPTG/X-Gal plates after incubation at 37°C overnight. Then,plasmids were extracted and sequenced from five positive colonies.

Table 1 Background information on the five Chinese Caprine arthritis-encephalitis virus isolates analyzed in this study

Table 2 Primers for the amplification of the whole genomes of Caprine arthritis-encephalitis virus isolates
To assemble the CAEV genomic sequences,seven amplified segments were sequentially assembled end to end,and then,the obtained genome sequences were annotated by comparison with the reference genomes published in GenBank.
2.3.Genotyping
The five CAEV genome sequences were successfully amplified. Because the strains showed homology,five distinctive sequences among them were aligned with 24 SRLV partialgagandgag-polsequences from GenBank,including CAEV isolates from other parts of the world,which were representative of eight sub-genotypes of the four known SRLV genotypes. For a robust and accurate phylogenetic analysis of the partialgaggene andgagpolsequences,the maximum-likelihood-based trees were constructed using MEGA X Software (Kumaret al.2018),and the Kimura 2-parameter model plus gamma distribution (K2+G) was chosen as the best model for nucleotide substitution. The reliability of the phylogenetic relationships was evaluated using a nonparametric bootstrap analysis with 1 000 replicates.
2.4.Whole-genome sequences and encoded protein similarity analyses
Whole genomes and encoded protein similarity matrices were constructed for whole-genome sequences and theenv,gag,pol,rev,vprandvifgenes of known CAEVs using Sequence Demarcation Tool v1.2 (Muhireet al.2014).
2.5.Phylogenetic analyses
The sequences were aligned with sequences of other CAEV (Appendix A). A multiple sequence alignment was performed using MAFFT version 7 (https://mafft.cbrc.jp/alignment/server/) (Katohet al.2002). The alignment was manually checked and end-trimmed to match the obtainedenv,gag,pol,rev,vprandvifgenes. The final multiple sequence alignment was used for the maximumlikelihood-based phylogenetic analyses with GTR+G+I(env,polandvifgenes),GTR+G (gagandrevgenes),TN93+G+I (vprgene) as the best-fit model of nucleotide substitution and a 1 000 bootstrap resampling using MEGA X (Kumaret al.2018).
2.6.Population structure
The CAEV population structure were analyzed using the program STRUCTURE Software v.2.3.4 (http://web.stanford.edu/group/pritchardlab/structure_software/release_versions/v2.3.4/html/structure.html),which applies a Bayesian statistical model to cluster genotypes into populations without prior information about their genetic relatedness,and in which the whole population is divided into K subpopulations characterized by a set of allele frequencies at each locus (Falushet al.2003). To run STRUCTURE,map distances were set equal to the parsimony-informative (PI) sites physical distances. The optimal number of populations was determined by running the model for K values from 2 to 6. For each K,10 runs were performed with MCMC run lengths of 200 000 and 1 200 000 burn-ins. Evanno’s method (Evannoet al.2005)to determine the trend of (LnPr(X|K),and STRUCTURE documentation (Janeset al.2017) were used to select the optimal K with STRUCTURE HARVESTER (Earl and Vonholdt 2012). The results of independent runs were merged by permutating clusters using StructureSelector(Li and Liu 2018) to generate a Q-value matrix. To evaluate the contribution of the ancestral component to each PI site,a run with the optimalKwas performed with the SITEBYSITE option selected.
3.Results
3.1.PCR amplification of CAEV whole genomes
We amplified seven segments of approximately 450,1 700,1 000,900,2 500,1 500 and 2 200 bp,respectively(Fig.1). The amplified fragments were then cloned and sequenced using Sanger sequencing methods.Additionally,the obtained sequences were assembled using DNAMAN 8.0,and the genomes of 239,CGu-3,CSI-4,gs-35V and SH-1 were 9 172,9 118,9 138,9 171 and 9 163 bp,respectively. These genome sequences were annotated using Sequin Software. Then,the viral genome sequences were submitted and deposited in GenBank under accession nos.KT214469.1,KT749878.1,KT749879.1,KT749880.1 and KT749881.1,respectively.
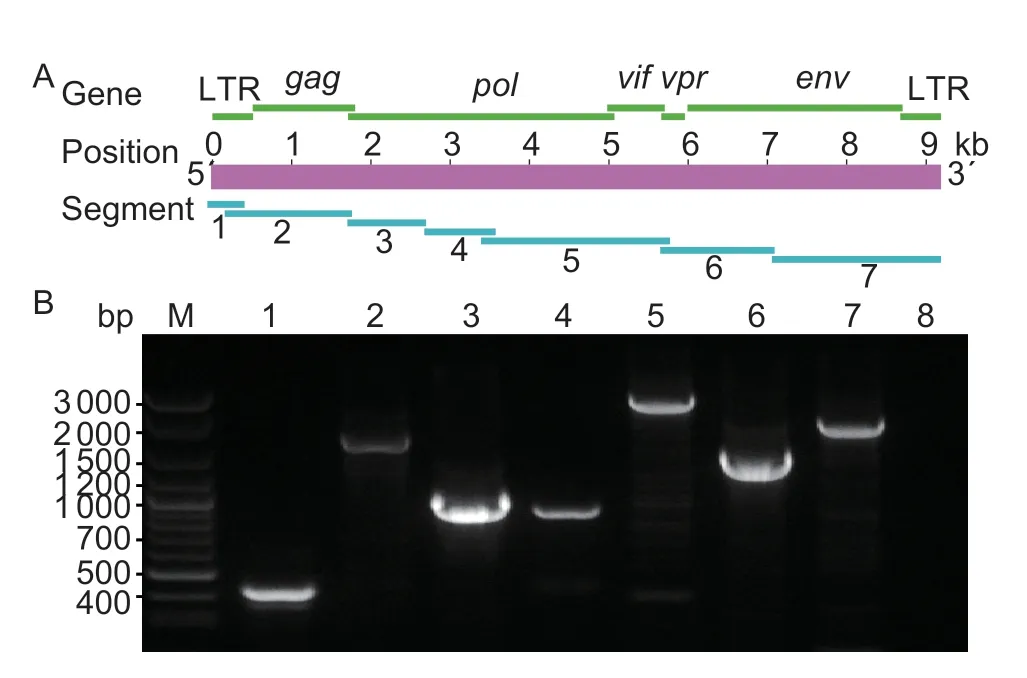
Fig.1 Whole genome amplifications of Chinese Caprine arthritis-encephalitis virus (CAEV) isolates. A,schematic diagram of the CAEV genome. Green bands indicate the position of encoded genes. Purple band indicates the length of CAEV genome. Blue band indicate the position of amplification segments. B,amplification result of seven segments of CAEV genome. M,DNA marker;1-7,segments 1-7;8,blank control.
3.2.Genotype identification
After sequencing,two genotyping methods based on the partialgag(684 bp) andgag-pol(1.8 kb) genes were selected. Gene sequences of several closely related SRLV genotypes and subgenotypes were downloaded from the GenBank database. A phylogenetic analysis was performed to align these five sequences with other reference sequences,as shown in Fig.2-A and B. The Chinese strains 239,CGu-3,CSI-4,gs-35V and SH-1 were all grouped into the SRLV subgenotype B1 branch in the phylogenetic tree,indicating that the earliest Chinese strain had only one subgenotype.
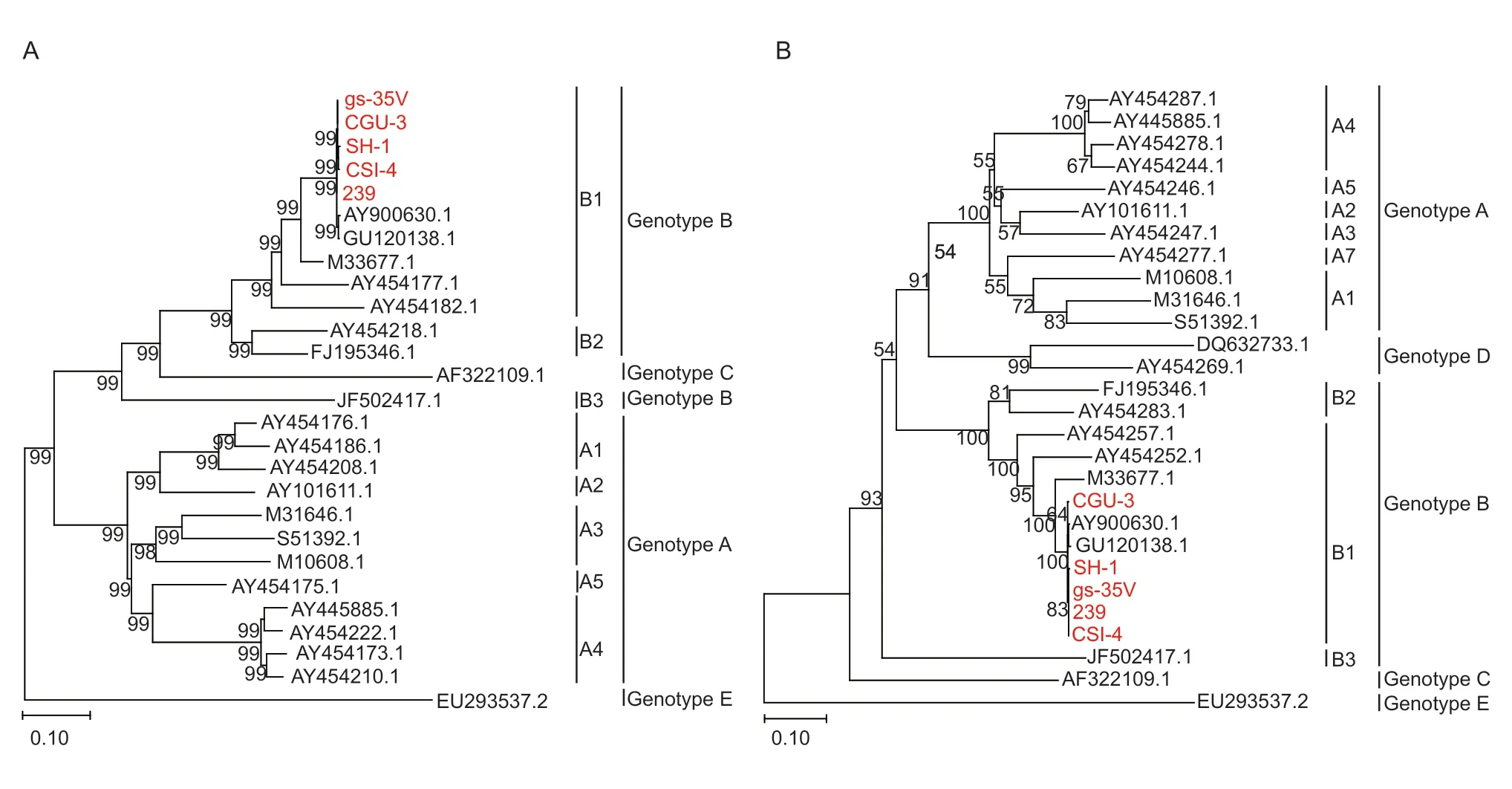
Fig.2 A genotypic analysis of Caprine arthritis-encephalitis virus (CAEV) isolates in China using the maximum-likelihood (ML)method. The evolutionary history was inferred using the ML method and the Kimura 2-parameter model (Kimura 1980). ML trees constructed based on a 684-bp fragment of the partial gag gene (A) and a 1 133-bp fragment of the 1.2-kb gag-pol gene (B). The CAEV genomes sequenced in this study are marked in red.
3.3.Similarity analysis
The sequences showed 97-100% identities with two other CAEV strains,Gansu and Shanxi,isolated from dairy goats in China (Fig.3). Additionally,when compared with CAEV strains from outside of China,it showed 58-93%identity levels. The similarities of six encoded protein are shown in Appendices B-G,and the sequences showed 98.0-99.9%,98.6-100%,99.0-99.8%,97.7-100%,98.9-100% and 99.6-100% identities to the Env,Gag,Pol,Rev,Vpr-like and Vif proteins of Chinese CAEV strains,respectively,but 51.4-81.7%,65.1-92.7%,71.7-92.1%,9.3-88.4%,63.9-89.7% and 49.1-88.2% identities to the Env,Gag,Pol,Rev,Vpr-like and Vif proteins from outside of China,indicating the high amino acid and genome similarities among Chinese strains.
3.4.Phylogenetic analyses
The phylogenetic trees constructed based onenv,gag,pol,rev,vprandvifgenes are shown in Fig.3. These strains exhibited close relationships with two other Chinese CAEV strains,Gansu and Shanxi (GenBank accession nos.AY900630.1 and GU120138.1,respectively),forming a distinct lineage in accordance with their geographical distribution (Fig.4-A-F),and the results also indicated that these seven Chinese CAEV strains (Gansu,Shanxi,239,CGu-3,CSI-4,gs-35V and SH-1) clustered with GenBank reference strain Clements (GenBank accession no.NC_001463.1),indicating that they seem to have a common ancestor.
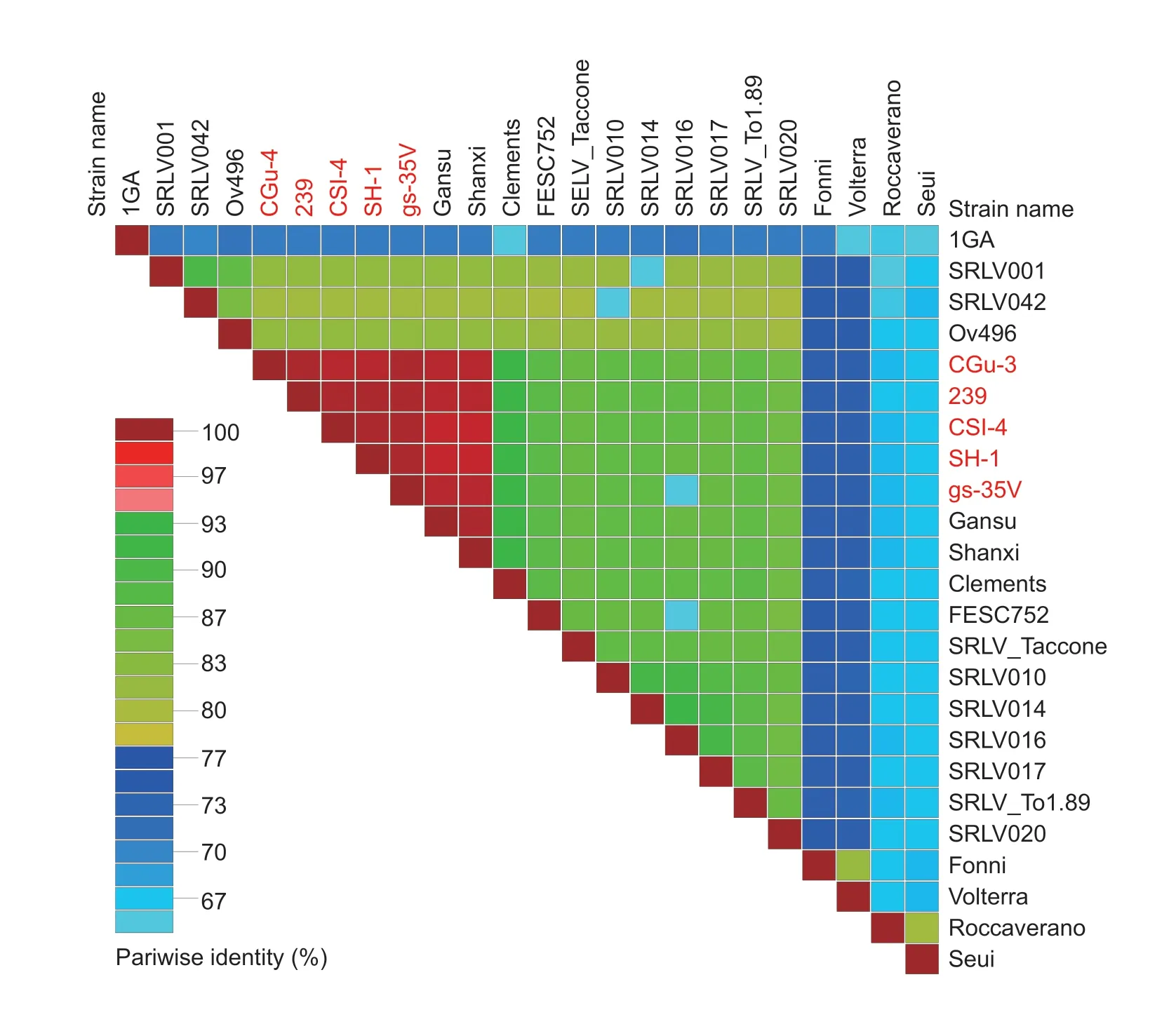
Fig.3 A pairwise whole-genome identity matrix of Caprine arthritis-encephalitis virus (CAEV) including strains isolated in China.The CAEV genomes sequenced in this study are marked in red.

Fig.4 Phylogenetic trees constructed based on six genes,env (A),gag (B),pol (C),rev (D),vpr (E) and vif (F) genes,using MEGA X.The Caprine arthritis-encephalitis virus (CAEV) genomes sequenced in this study are marked in red.
3.5.Population structure
To estimate the optimal number of subpopulations in the CAEV dataset,STRUCTURE was run for values ofKfrom 2 to 6. The CAEV yielded a major peak atK=3 (Appendix H),indicating the optimal subpopulation number was 3. Consequently,an analysis of ancestry components was performed forK=3,and genomes were plotted in accordance with their geographical origins (Fig.5). The clustering trend of the ancestral population included three major components and was highly site specific(e.g.,population 1 occurs in China,whereas population 3 occurs in Italy and Norway). Population 1 for Chinese strains included the CAEV isolates from the provinces of Gansu,Guizhou,Shaanxi,Shandong and Sichuan,indicating that this population had a unique major ancestry component that differed from those of other strains.
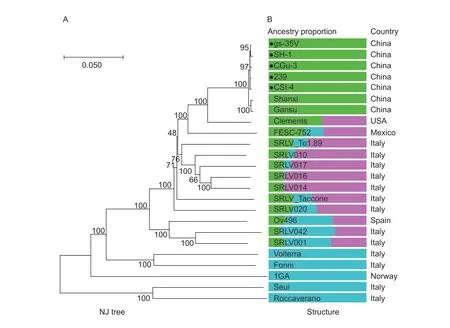
Fig.5 Population structure of Caprine arthritis-encephalitis virus (CAEV) constructed based on complete-genome sequences published in GenBank. Bar plot representing the proportion of ancestral population components from the STRUCTURE linkage model for K=3 based on whole-genome sequences. Each column represents a CAEV genome,and the virus name is marked in the column. Genomes are ordered by population. The phylogenetic tree was constructed using MEGA X. The CAEV genomes sequenced in this study are marked with black solid circles.
4.Discussion
Caprine arthritis-encephalitis has been recognized as an imported animal infectious disease in China,and its recessive and latent infections cause a health risk to the goat breeding industry (Baihuojiaet al.1989;Huet al.1989;Liet al.1990;Linet al.1990;Wanget al.1994). In this work,we selected five CAEV strains isolated from five Chinese provinces (Gansu,Guizhou,Shaanxi,Shandong and Sichuan) that were isolated between 1989 and 1994,and we performed cloning and sequencing of the wholegenome sequences of these five viruses,which provide genomic evidence for the source-tracing and control of CAEV in China.
The current SRLV phylogeny consists of five genotypes that are further divided into more than 20 subgenotypes,which emphasizes the high genetic variation among SRLV strains (Michielset al.2020;Molaeeet al.2020).Fortunately,only subgenotype B1 was found in China,indicating that the Chinese circulating strains were less complicated than those in other countries,such as Italy.However,Caprine arthritis-encephalitis is an under-studied animal disease in China,and no eradication programs nor existing diagnostic tests are available. Thus,our study provides insights into CAEV circulation and genetically characterized the current Chinese strains present in naturally infected goats.
A sequence analysis of the whole genomes of five Chinese strains showed that these viruses were closely related to SRLV subgenotype B1,indicating a common ancestor. The phylogenetic tree had the same shape as the tree constructed based on all six genes,e.g.,env,gag,pol,rev,vprandvif,suggesting the common evolution of Chinese CAEV strains. By estimating the drift level from a hypothetical common population,the applied STRUCTURE model allows inferences to be made about the most likely original location of the sampled genomes.Overall,our data indicated that CAEV lineages diverged from an ancestral population circulating in China (within one major ancestral component),USA/Mexico/Italy/Spain(within two or three ancestral population components)and Italy/Norway (within one major ancestral population component),indicating the regional distribution of CAEV.The ancestral population component of the whole genomes of the five Chinese strains in this study was shared with two other Chinese strains,Gansu and Shanxi,but was not detected in other strains,indicating that this ancestral lineage may be less common in other countries.
When combined,our results indicated a single subgenotype,B1,and a low seroprevalence for CAEV in China (Sunet al.2018),which indicated that implementing eradication programs to eradicate Caprine arthritisencephalitis would be less complicated than in Europe.In particular after the African swine fever outbreak and epidemic in China,the Chinese authorities restricted the cross-regional transportation of livestock (http://www.moa.gov.cn/ztzl/fzzwfk/zcfg/201809/t20180925_6158480.htm).Therefore,setting standards for a CAEV-free zone,and implementing a CAEV-free zone policy in some regions of China,would be beneficial for achieving a Caprine arthritis-encephalitis-free country.
Previous studies in China speculated that the Chinese CAEV strains originated from Saanen dairy goats and Toggenburg goats imported in the 1980s from the United Kingdom,but our results did not find direct genomic evidence to support this conclusion. This might be because: 1) the number of sequenced CAEV genomes worldwide is less than 30,and there are especially limited CAEV genome sequences from the United Kingdom;and 2) the population structure showed a unique pattern of ancestral population components. Therefore,we cannot exclude the possibility of CAEV from local goats or sheep being transmitted to the imported Saanen dairy goats and Toggenburg goats. To address the above issues,it will be necessary to conduct a more detailed molecular epidemiological investigation in China and obtain more genome sequences. In addition,increasing the numbers of British CAEV strains would be beneficial to the traceability.
5.Conclusion
The whole genomes of Chinese CAEV strains isolated in 1989-1994 confirmed that they belonged to subgenotype B1 and contained unique ancestral components,sharing 79.5 to 86.9% identities with strains from others countries.Additionally,the Chinese CAEV strains isolated from different provinces over five years were still highly homologous. The results of this study establish a basis for performing further epidemiological investigations and implementing CAEV control strategies in China.
Acknowledgements
This work was funded by the National Key Research and Development Program of China (2016YFD0500908). We are indebted to Profs.Wang Zhicai,Baihuojia Huermaxi,Lin Jie,Hu Zeyuan and other researchers participated in CAEV isolation and identification during 1985 and 1994 who worked at the Institute of Veterinary Medicine,Xinjiang Academy of Animal Sciences,China,for providing CAEV strains for this study.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Ethical approval
This study does not contain any studies with animal subjects performed by any authors.
Appendicesassociated with this paper are available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
 Journal of Integrative Agriculture2023年3期
Journal of Integrative Agriculture2023年3期
- Journal of Integrative Agriculture的其它文章
- Germinated brown rice relieves hyperlipidemia by alleviating gut microbiota dysbiosis
- Melatonin treatment alleviates chilling injury in mango fruit 'Keitt'by modulating proline metabolism under chilling stress
- Changes in the activities of key enzymes and the abundance of functional genes involved in nitrogen transformation in rice rhizosphere soil under different aerated conditions
- Growth and nitrogen productivity of drip-irrigated winter wheat under different nitrogen fertigation strategies in the North China Plain
- Effect of fertigation frequency on soil nitrogen distribution and tomato yield under alternate partial root-zone drip irrigation
- Phylogenetic and epidemiological characteristics of H9N2 avian influenza viruses in Shandong Province,China from 2019 to 2021