
Impaired autophagy in amyloid-beta pathology: A traditional review of recent Alzheimer's research
2023-02-06 08:07:50MinghaoYuanYangyangWangZhentingHuangFengJingPeifengQiaoQianZouJingLiZhiyouCai
Minghao Yuan, Yangyang Wang, Zhenting Huang, Feng Jing, Peifeng Qiao, Qian Zou,Jing Li, Zhiyou Cai,?
1Chongqing Medical University, Chongqing 400042, China;
2Department of Neurology, Chongqing General Hospital, University of Chinese Academy of Sciences, Chongqing 400013, China;
3Department of Neurology, Chongqing School, University of Chinese Academy of Sciences, Chongqing 400013, China;
4Chongqing Key Laboratory of Neurodegenerative Diseases, Chongqing 400013, China.
Abstract Alzheimer's disease (AD) is the most prevalent neurodegenerative disorder. The major pathological changes in AD progression are the generation and accumulation of amyloid-beta (Aβ) peptides as well as the presence of abnormally hyperphosphorylated tau proteins in the brain. Autophagy is a conserved degradation pathway that eliminates abnormal protein aggregates and damaged organelles. Previous studies have suggested that autophagy plays a key role in the production and clearance of Aβ peptides to maintain a steady-state of Aβ peptides levels.However, a growing body of evidence suggests that autophagy is significantly impaired in the pathogenesis of AD, especially in Aβ metabolism. Therefore, this article reviews the latest studies concerning the mechanisms of autophagy, the metabolism of Aβ peptides, and the defective autophagy in the production and clearance of Aβ peptides. Here, we also summarize the established and new strategies for targeting autophagy in vivo and through clinical AD trials to identify gaps in our knowledge and to generate further questions.
Keywords: Alzheimer's disease, autophagy, amyloid-beta, amyloid precursor protein secretases, metabolism
Introduction
Alzheimer's disease (AD) is an age-related neurodegenerative disease that is characterized by extensive memory loss, ongoing cognitive dysfunction, and behavioural disorders[1–3]. Global cost of AD is estimated to be 818 billion US dollars, which puts a huge strain on political economy[4]. The number of people diagnosed with AD is also expected to rise from 46.8 million in 2015 to 74.7 million by 2030.This trajectory suggests that this figure will rise to 131.5 million by 2050[4], if we do not develop effective interventions. Unfortunately, AD begins to develop more than 10 years before symptoms begin to manifest[5]. Research to date has suggested that various alterations, including the production and accumulation of amyloid-beta (Aβ) and phosphorylated tau proteins, as well as mitochondrial and synaptic damage, inflammatory responses,oxidative stress, hormonal imbalance, and neuronal loss, are involved in the AD progression[6–14]. These pathological changes are persistent in Aβ peptide aggregation that in turn forms senile plaques and drives the accumulation of hyperphosphorylated tau proteins. This creates neurofibrillary tangles that in turn impairs nervous system function[1,15–19]and accelerates deterioration. Therefore, understanding these mechanisms could provide insights into the development and progression of AD as well as possible therapeutic targets.
Autophagy is a self-cannibalisation process that involves the decomposition of cell structures through lysosomes, maintaining a balance in both synthesis,degradation, and cellular circulation[20–22]. Autophagy can be both selective or non-selective, depending on the properties of specific target proteins[23]. In most cases, autophagy refers to macroautophagy that is an evolutionarily conserved catabolic process involved in vesicle formations,i.e., autophagosomes, which engulf cellular macromolecules and organelles. The tightly coordinated multi-step process involved in autophagy is associated with protein products from autophagy-associated genes, which adhere to the target and participate in the entire cycle[24]. Under normal circumstances, autophagy plays a type of"housekeeping" role in cellular homeostasis by removing denatured or misfolded proteins and damaged organelles as we age. Induced by various stress signals including certain nutrients, hypoxia, and reactive oxygen species[13,25], soluble proteins and organelles in the cytoplasm are degraded into amino acids to supply energy and for biosynthesis.
Aβ has also been found to have a continuously toxic effect on neurons and synaptic facilitation[16].However, while impaired autophagy has previously been identified in AD[26–29], only recently has autophagy been found to mediate Aβ secretion into extracellular space[27]. In an AD brain,autophagosomes and late autophagic vacuoles accumulate in dystrophic neurites, suggesting that there is underlying autophagy impairment[27,29].Additionally, the selective accumulation of lysosomal dense bodies in the brains of AD cases implies that there is abnormal autolysosomal proteolysis[30–31].However, the precise alterations in autophagy processes in AD remain unclear.
This review highlights impaired autophagy in the pathogenesis of AD. We initially describe Aβ metabolization, the pathway of autophagy, and the role of autophagy in neurodegenerative diseases. This is followed by a comprehensive summary of defective autophagy in promoting an Aβ precursor protein(APP) processing that leads to an enhanced activity or increased levels of the beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) and γ-secretase.This again accelerates the production of Aβ into a state of overproduction. We then discuss the association between defective autophagy and Aβ clearance failures. In addition, we provide a summary of compounds and drugs that target autophagy in AD animal models or AD patients.
Autophagy
Autophagy is a natural self-degradation process that promotes cell survival in response to nutritional stress that can be caused by starvation and by certain diseases. Autophagy also balances sources of cellular building materials and energy intake. The literal meaning of autophagy is 'self-eating', as 'auto' meansselfand 'phagy' meanstoeat. During this aptly named"self-eating" process, cells remove unwanted molecules and dysfunctional cellular components. The process is divided into macroautophagy,microautophagy and chaperone-mediated autophagy according to the mechanism through which intracellular materials are delivered into lysosomes for degradation[20]. The different types of autophagy are illustrated inFig. 1and described in detail below.
Macroautophagy starts with the formation of a double-membraned vesicles called autophagosomes[32–33], and then cytoplasmic components, such as organelles and proteins, are enveloped by doublemembrane vesicles[33]. The outer membrane of the autophagosome then fuses with lysosomes to form autolysosomes, where organelles and proteins are degraded by lysosomal acid hydrolase. In microautophagy, lysosomes directly degrade cytoplasmic components that are engulfed by invagination of the membrane[20]. It is generally accepted that microautophagy can be either nonselective or selective[32]. Selective autophagy includes mitophagy, pexophagy, reticulophagy, ribophagy,lipophagy, xenophagy, nucleophagy and ferritinophagy[34], which can target specific tissues,malignant cells, damaged organelles, aggregated proteins, invasive pathogens, and excessive peroxisomes[35]. It is achieved by the autophagy receptor that adheres to the targetviathe autophagyrelated protein 8 (ATG8) family on the autophagosome membrane. Then poly-ubiquitin on the target is recognized by receptor proteins, which initiates selective autophagy. In chaperone-mediated autophagy, cytoplasmic proteins are degraded in lysosomes or vacuoles after being transported by molecular chaperones, such as heat shock cognate protein 70[23](Fig. 1).
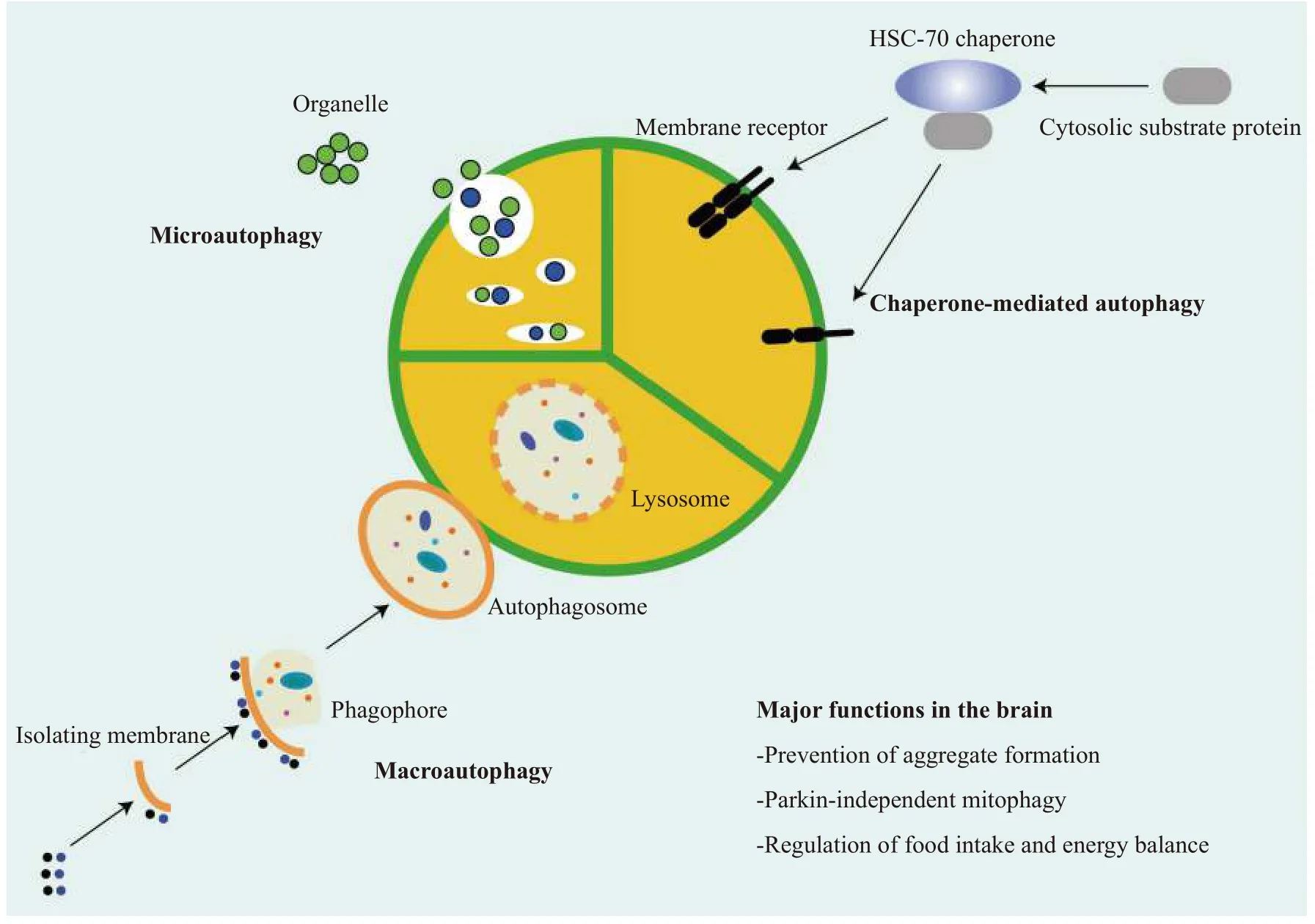
Fig. 1 An overview of autophagy. Autophagy is mainly divided into three types, depending on the way in which materials are transported to the lysosome: 1) Macroautophagy, the endoplasmic reticulum-derived membrane wraps around the substrate to form an autophagosome,which then fuses with lysosomes and degrades its contents; 2) Microautophagy, the membrane of the lysosome directly wraps proteins into endochylema and the proteins are the degraded in then lysosome; and 3) Chaperone-mediated autophagy, target proteins bound to a chaperone, such as heat shock cognate 70 (HSC-70) are transported to the lysosome and then degraded by hydrolase.
In summary, autophagy is a highly conserved pathway that degrades large proteins and organelles by lysosomes (Fig. 1), which plays a vital role in the circulation, infection, immunity, and metabolism of cells[36–37]. Under normal developmental conditions,cells perform low levels of autophagy, specifically basal autophagy, to maintain cellular homoeostasis.However, under various stressors, autophagy becomes highly dynamic, and its progress can be split into initiation, nucleation, elongation, closure, and lysosomal fusion[38–41](Fig. 2).
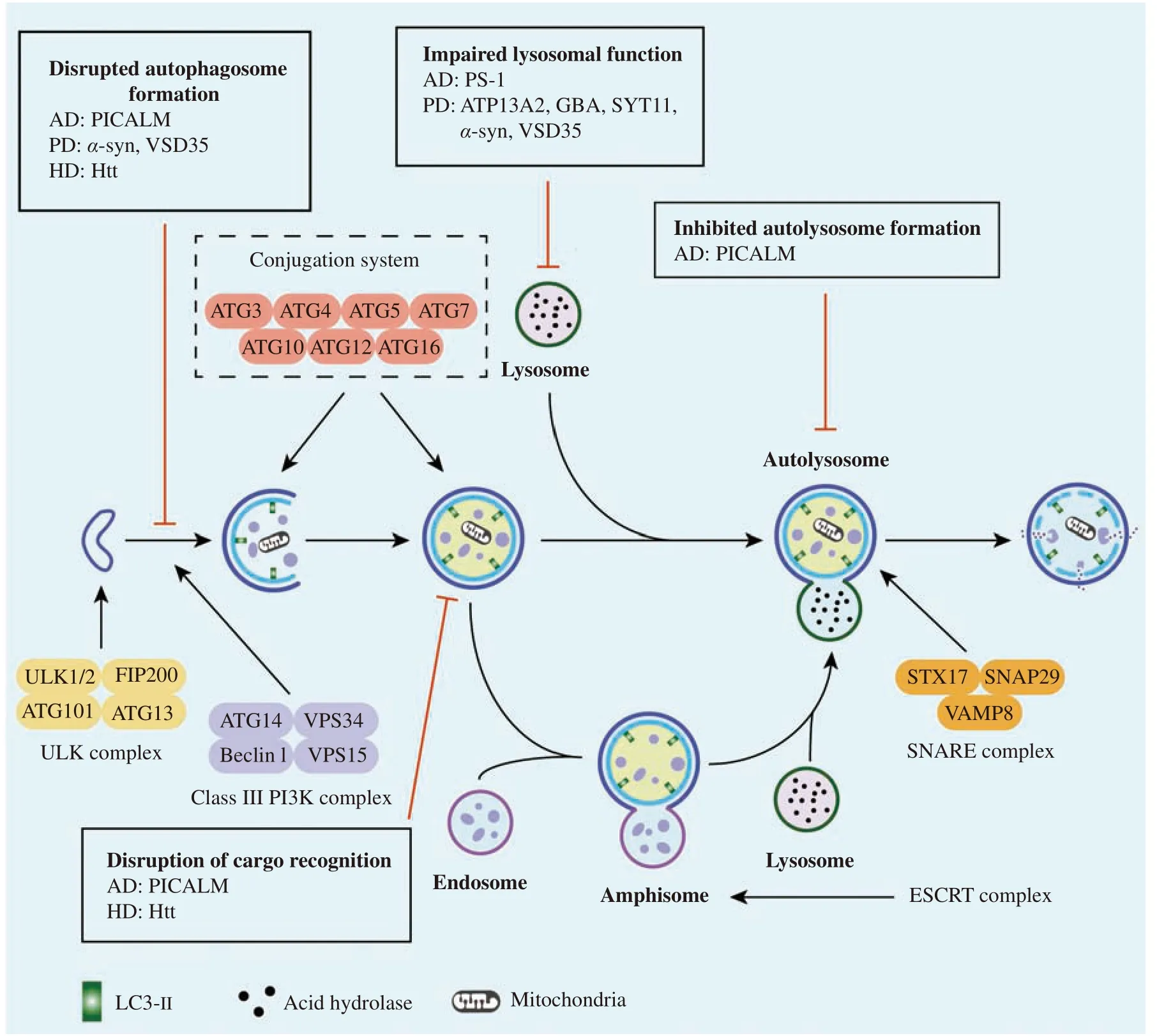
Fig. 2 The general processes involved in autophagy and impaired autophagy in neurodegenerative disorders. There are five steps involved in autophagy, i.e., initiation, elongation, closure, fusion, and degradation. Several important factors, such as ULK complex, class ⅢPI3K complex, conjugation system, ESCRT complex, and SNARE complex, play vital roles in the form of autolysosome. In addition, the amphisome or autophagosome fuses with the lysosome to form the autolysosome, finally eliminating cargos. However, various processes of autophagy are disrupted in neurodegenerative diseases, such as Alzheimer's, Parkinson's, and Huntington's diseases. α-syn: α-synuclein; AD:Alzheimer's disease; ATG: autophagy related gene; ATP13A2: ATPase type 13A2; ESCRT: endosomal sorting complex required for transport; FIP200: FAK family kinase-interacting protein of 200 kDa; GBA: glucocerebrosidase; HD: Huntington disease; PD: Parkinson disease; PICALM: phosphatidylinositol binding clathrin assembly protein; PS-1: presenlin1; SNAP29: neuronal synaptosome associated protein 29; SNARE: soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins; STX: syntaxin; ULK1: unc-51-like kinase 1; VAMP8: vesicle-associated membrane protein 8; VPS: vacuolar protein sorting.
Initiation
The activity of the unc-51-like kinase 1 (ULK1)complex (comprising ULK1/ATG1, ATG13, FAK family kinase-interacting protein of 200 kDa, and ATG101) is required for the initiation of autophagy[42].The ULK1 complex is affected by certain stress signaling pathways, such as the mammalian target of the rapamycin (mTOR) pathway[43].
Nucleation
Nucleation of a double-membrane structure called the phagophore requires a class Ⅲ phosphatidylinositol 3-kinase complex. This complex consists of vacuolar protein sorting 34 (VPS34), VPS15, ATG14,and Beclin1 and generates phosphatidylinositol-3 phosphate, which is essential for nucleation of vesicles[44].
Elongation and closure
The elongation and closure required to form autophagosomes depend on two ubiquitin-like conjugation systems, known as the ATG12-ATG5 system and the ATG8/LC3-lipid phosphatidylethanolamine (PE) system[45–47]. These systems interact and regulate each other with the participation of ubiquitin activases (E1) and ubiquitin conjugases (E2). ATG12,activated by ATG7, combines with ATG5 through transport of ATG10 and then binds to ATG16 to form a multi-body complex of ATG12-ATG5-ATG16[44].This complex is located on the surface of the outer membrane of the vacuole and participates in the elongation of this membrane. LC3-Ⅰ, which is mediated by ATG7 and ATG3, conjugates with PE to form LC3-Ⅱ to participate in membrane elongation[45].LC3-Ⅱ, commonly regarded as a marker of autophagy, is also an important signaling regulatory protein located on the membrane of autophagic vacuoles.
Lysosomal fusion
A fusion of lysosomes with autophagosomes to form an autolysosome is the final step in autophagy[45].As specialized organelles, lysosomes play an important role in breaking down extracellular materials and recycling cellular components in different pathways[48]. The fusion between autophagosomes and lysosomes requires a soluble Nethylmaleimide-sensitive factor activating the protein receptor complex that comprises syntaxin 17,synaptosomal-associated protein 29, and vesicleassociated membrane protein 8[49].
Formation of amphisomes
An autophagosome can also fuse with a late endosome (LE) to form an amphisome that contains markers of both autophagosomes (LC3) and endosomes (ras-related protein in brain 5 [RAB5],RAB7, and RAB11)[50]. Members of the endosomal sorting complex required for complex transport play key roles in formation of amphisomes and autolysosomes during autophagy[51].
Degradation and reuse
The inner membranes and components of autolysosomes are degraded by lysosomal hydrolases,and a series of lysosomal proteases (e.g., cathepsins B,D, and L) degrade the contents in mammalian cells.When macromolecules have been degraded in lysosomes, monomer molecules, such as amino acids and lipids, are recycled into the cytoplasm for reuse;however, little is known about what happens in this stage. Atg22 is essential for the reuse in yeast, but no counterparts of Atg22 have been found in mammals[52]. Additionally, little is known about the role of autophagy in the reuse of large molecules, such as carbohydrates and lipids.
Amyloid-beta metabolism
The two hallmark pathological changes, which are required for a diagnosis of AD, are deposition of the extracellular Aβ plaque and neurofibrillary tangles comprised of microtubule-binding protein tau.Following a decade of research, the excessive production and impaired Aβ clearance are now known to cause these amyloid plaques and deleterious cascades involved in the pathogenesis of AD.
Amyloid-beta production
Aβ is produced by way of sequential cleavage of the APP by β- and γ-secretases[53]. APP, which is a type Ⅰ transmembrane protein, exists in various tissues and is intensively expressed on the membrane of neuronal synapses[54]. There are two main metabolic pathways for APP: a non-amyloidogenic and an amyloidogenic pathways (Fig. 3). The nonamyloidogenic pathway is mediated by α- and γsecretases, soluble Aβ precursor protein-alpha(sAPPα), p3 peptides (Aβ17–40and Aβ17–42), and the APP intracellular domain, but not toxic Aβ peptides,are produced, because the cutting site of α-secretase is inside the Aβ sequence[55–56]. Whereas, in the amyloidogenic pathway, Aβ is produced and released by a combination of BACE1 and γ-secretase in the intracellular compartments, including the trans-Golgi network, endosomes, and autophagosomes[57]. Aβ40 and Aβ42 are the most common peptides of Aβ subtypes. In comparison with Aβ40, Aβ42 is more hydrophobic and prone to aggregation, forming oligomers or fibrils and finally senile plaques, which is one of the main histological features of AD[57–60].However, there is no evidence of an association between the number of senile plaques and the severity or progression of AD[57].
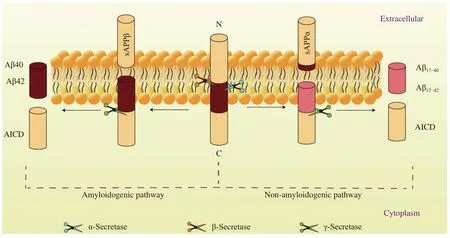
Fig. 3 Amyloid-beta precursor protein processing. In amyloidogenic pathway, amyloid-β precursor protein is cleaved by β- and γsecretases to produce Aβ40 and Aβ42. In non-amyloidogenic pathway, amyloid-β precursor protein is cleaved by α- and γ-secretases to produce Aβ17–40 and Aβ17–42, which are not neurovirulent. Aβ: amyloid-beta; AICD: β-amyloid precursor protein intracellular domain; APP:β-amyloid precursor protein.

Fig. 4 Amyloid-beta transport at the BBB is regulated by RAGE and LRP-1. LRP-1 regulates Aβ transporting from the brain parenchyma to peripheral circulation across the blood-brain barrier (BBB), while RAGE regulates Aβ transporting from peripheral circulation to brain parenchyma across the BBB. Aβ: amyloid-beta; LRP-1: low density lipoprotein receptor-related protein 1; RAGE:receptor for advanced end glycation products.
Due to different roles the of α-, β-, and γ-secretases in the metabolism of APP, the products they produce,when cleaving APP, have different cellular effects.The α-secretase enzyme plays a major role in the production of sAPPα. Upregulated α-secretase enzyme can promote the production of sAPPα and reduce production of Aβ, whereas the β-secretase acts as a rate-limiting enzyme in the formation of Aβ. The γsecretase enzyme plays a pivotal role in catalysing the production of Aβ in the metabolism of APP and in determining the terminal stage of Aβ. In general,upregulation of α-secretase activity and inhibition of β- and γ-secretases are important for reducing Aβ formation.
Amyloid-beta clearance
In the brain, neprilysin and insulin-degrading enzyme are considered to be the main enzymes involved in Aβ degradation[61]. Neprilysin, a type Ⅱmetalloproteinase, is responsible for extracellular degradation of Aβ42, and its activation can reduce the aggregation and toxicity related to Aβ, leading potentially to cognitive improvements[58,62]. The insulin-degrading enzyme is a mercaptan metalloproteinase with an important role in degrading soluble Aβ monomer and reducing the deposition and accumulation of Aβ[58,60]. Additionally, M2 microglia and astrocytes can clear Aβ through phagocytosis.Apolipoprotein E and low-density lipoprotein receptor-related protein 1 (LRP-1) have been demonstrated to assist astrocytes with degradation[56,60]. Moreover, LRP-1 can mediate the transport of Aβ across the blood-brain barrier (BBB)to the peripheral blood circulation[56,63–64], and brain microvascular endothelial cells at the BBB participate in the clearance of AβviaLRP-1 and Pglycoprotein[63,65].
Amyloid-beta in Alzheimer's disease
For some time, it has been assumed that an imbalance between the production and clearance of Aβ leads to its accumulation in the brain. Normally, a dynamic balance is maintained between the generation and decomposition of Aβ in the brain. However, when various pathogenic factors are present, the Aβ metabolism is disrupted, causing its accumulation that affects neurological function[66]. In the early stage of AD, extensive aggregation and accumulation of Aβ may occur in the brain, leading to pathological changes, including impaired autophagy[67],apoptosis[68], and oxidative stress[69]as well as abnormal growth of axons[70].
In addition, abnormal accumulation of Aβ is known to be an initial factor in the inflammatory response observed in AD. Senile plaques formed by Aβ promote an inflammatory response, causing proliferation of glia[71]. Accumulated Aβ binds to the receptor on microglial cell membranes and facilitates secretion of inflammatory factors from microglia,inducing an inflammatory response, whereas activated inflammatory factors impair normal neurons nearby[72–74]. Furthermore, a recent study has indicated that microglia activated by Aβ plaques promotes the propagation Aβ into unaffected brain regions[75]. It is therefore reasonable to suggest that the Aβ pathology may interact with pathological changes in the brain,causing a vicious circle that eventually accelerates the progression of AD.
Impaired autophagy induces overgeneration of amyloid-beta
Aβ peptides are generated from APP through sequential cleavage by BACE1 and the γ-secretase complex[54,56]. A recent study has indicated that pathological Rab5 overactivation during AD induces endosomal dysfunction, which disrupts the formation of autolysosomes, eventually causing autophagy dysfunction[76]. Additionally, a detective autophagy,characterized by a deficit in autolysosome acidification in neurons, has been found to occur before extracellular amyloid deposition in five AD mice models[77]. Given the importance of autophagy in Aβ metabolism, it is possible that an impaired autophagy enhances activities or levels of the βsecretase and γ-secretase, resulting in Aβ overproduction.
Impaired autophagy and BACE1 in Alzheimer's disease
BACE1 is a key enzyme in the amyloidogenic pathway of APP and is responsible for regulating the degradation of APP to produce Aβ. BACE1 is a transmembrane aspartic protease that is highly expressed in brain neurons. There is a compelling evidence for an age-associated increased activity of BACE1 in the AD brain[78]. As a result, much research has focused on BACE1 as a target for the treatment and prevention of AD[79–81]. BACE1 is initially synthesized into a protein precursor in the endoplasmic reticulum, before being transported to the Golgi body, where it is glycosylated and modified into a mature BACE1 protein. Mature BACE1 is then internalized through the plasma membrane or transferred from the trans-Golgi network directly to the endosome, providing a suitable acidic environment for the protein's activity[82–84]. Eventually, BACE1 is degraded within the lysosomes.
Previous studies have indicated that autophagosomes are constantly produced in the distal axons and move retrogradely to the soma for lysosomal proteolysis[85–89]. This transport is triggered by fusion of the nascent autophagosome with LE and driven by dynein-SNAPIN complexes with LE[88]. Interestingly,autophagosomes have been shown to transport BACE1, thereby regulating its trafficking and degradation[90]. Bothin vivoandin vitrostudies have shown that a large amount of BACE1 is recruited into autophagy vesicles and transported to the soma,augmenting the transport of BACE1 to lysosomes for degradation[90]. Therefore, autophagy is considered another pathway for trafficking of BACE1 for degradation in neurons. However, under pathological conditions, an AD-associated defective autophagy causes an impaired retrograde transport, which results in an accumulation of BACE1, enhancing the processing of APP by BACE1 in axons[91]. Increased BACE1 in distal axons augments production of Aβ in the APP process, exacerbating the AD pathological changes. Therefore, a potential therapeutic strategy that increases the induction of autophagy to improve the trafficking of BACE1 may be reasonable for treating the early stage of AD in the future.
In addition to regulating the trafficking and degradation of BACE1, the disruptions of upstream pathways, including phosphatidylinositol 3-kinase/Akt/mTOR and PPARγ/AMPK/mTOR, can indirectly decrease BACE1 levels by activating autophagy. mTORC1, which is a serine-threonine protein kinase, is a classical regulatory molecule for autophagy. This protein kinase inhibits autophagy by binding to the ULK1 complex and phosphorylating both ATG13 and ULK1[43]. However, an aberrant upregulation of the mTOR signaling was detected in AD brains[29,92–94], suggesting an enhanced mTOR activity and an inhibited autophagy during the AD process[95].Otherwise, several recent studies have shown that inhibition of the mTOR pathway can reduce BACE1 levels, decreasing production of Aβ[96–97]. These findings suggest that an elevated mTOR activity inhibits autophagy in AD, resulting in increased BACE1 levels and subsequently Aβ over-generation.
Impaired autophagy causes overactivation of the γsecretase
The protein γ-secretase complex includes presenilin, nicastrin, anterior pharynx defective-1, and presenilin enhancer 2, which plays a key role in the catabolic metabolization of APP. In cell experiments,an autophagy inhibitor (3-methyladenine) has been found to activate certain components of the γsecretase complex and significantly increase extracellular levels of Aβ42[98]. Furthermore, in AD patients, a defective autophagy can upregulate the expression of these components, thereby increasing the activity of the γ-secretase and promoting the Aβ production. Ohta Ket aldemonstrated that an impaired autophagy stimulated expression of PS1 and activated the γ-secretase[28]. When the autophagylysosomal system is impaired, the intracellular supply of amino acids decreases, because autophagy is required to maintain amino acid levels. Cellular amino acid deficiencies can lead to elevations in uncharged tRNA levels, which directly activate the environmental sensing protein, the general control non-derepressible 2(GCN2). As part of the amino acid imbalance, an increased GCN2 level causes phosphorylation of eIF2α that regulates the activating transcription factor 4. Finally, presenilin-1 is upregulated, and the γ-secretase is activated, leading to production of Aβ[28].
Impaired autophagy affects amyloid-beta clearance
Numerous studies have shown a potential association among autophagy, and Aβ clearance and deposition[27,99–105]. Large amounts of Aβ deposition interfere with the function of intracellular organelles,such as lysosomes, thereby increasing the accumulation of Aβ and promoting AD progression[104]. Aβ-derived diffusible ligands(ADDLs) can significantly reduce phosphorylated p70S6K expression, suggesting that the mTOR pathway inhibits an abnormal autophagy involved in an ADDL-induced autophagy[106]. Moreover, levels of Beclin-1, a key protein for initiation of autophagy, is significantly decreased in the early stage of AD,indicating that a reduced Beclin-1 level promotes neurodegeneration and accelerates the accumulation of Aβ[102–103]. Furthermore, autophagy also participates in the secretion of Aβviathe secretory pathway from the endoplasmic reticulum to the Golgi body and then to the plasma membrane or secretory lysosomal pathway[27,107]. Defective autophagy has been found to decrease the intracellular Aβ peptide load and accumulation of the extracellular Aβ in mice, which suggests that abnormal autophagy can reduce the degradation of Aβ, but also indicates that autophagy plays a role in secretion of Aβ[27]. Further studies on the dual roles of autophagy in the clearance and secretion of Aβ may contribute to a better understanding of the pathogenesis of AD.
Several recent studies have demonstrated that enhancement or activation of autophagy increases Aβ clearance and reduces its deposition. For example,Caccamoet aldemonstrated that an upregulation of p62 expression in an AD mouse modelviathe mTORdependent pathway, which activates autophagy,reduced level of Aβ and cognitive defects[99]. By contrast, Wanget al[108]found that the ratio of LC3B-Ⅱ/LC3B-Ⅰ increased in AD mice that received an oral rAAV/Aβ vaccination, and autophagy was enhanced with a decreased p62 level. However, the role of p62 in mediating autophagy to clear Aβ remains controversial and requires further investigation.
Rapamycin has been found to prevent an increase in calcium ions and to decrease the mitochondrial membrane potential in PC12 cells[109]. These alterations indicate that a moderate activation of autophagy can regulate a dynamic balance of calcium ions and maintain the stability of mitochondrial membrane potential, thereby alleviating the cytotoxicity induced by Aβ[109]. Additionally, mouse modelling research performed by Di Mecoet alshowed that after 12 weeks treatment with 12/15-lipoxygenase inhibitors, the level of Aβ was significantly decreased, and the effect was dependent on activation of autophagy in neurons[100].
In conjunction with these findings, investigators have also found that recovery of the autophagic flux can actually reverse the manifestations of AD caused by an accumulation of Aβ. The zinc ion carrier effect of hydroxychloroquine increases autophagic flux and reduces the accumulation of Aβ[105]. The epidermal growth factor receptor ErbB2 is thought to be dormant in the adult brain, but is activated in the hippocampus in AD patients. ErbB2 can dissociate Beclin-1 from the Vps34-Vps15 complex to inhibit the autophagic flux, suggesting that upregulated ErbB2 causes defective autophagy in AD[105]. When ErbB2 was downregulated in an AD mouse study, spatial learning and cognitive function appeared to be significantly improved, indicating that the downregulated ErbB2 can reverse inhibition of the autophagic flux and enhance the clearance of Aβ[110].
Generally, the intracellular domain of LRP-1 binds to phosphatidylinositol-binding clathrin assembly protein to regulate endosomal transcytosis of Aβ at the BBB[111–112](Fig. 4). However, cerebral accumulation of Aβ has been shown to accelerate autophagylysosomal degradation of LRP-1 in endothelial cells at the BBB, leading to dysfunction at the BBB[101].Unlike LRP-1, the receptor for advanced glycation end products (RAGE) transports Aβ from the systemic circulation into the brain. RAGE is thought to be an important factor mediating the cytotoxicity of Aβ and promoting AD pathogenesis. Moreover, RAGE has been reported to impair autophagy-lysosomal degradation, indirectly disrupting Aβ clearance[108].Further investigation has revealed that the Aβ oligomer causes impaired tight junction proteinsviaRAGE-mediated autophagy[113]. However, the precise relationship among autophagy, LRP-1, and RAGE in AD is not clear and requires further investigation.
Established and new strategies targeting autophagy in Alzheimer's disease
As has been previously discussed, Aβ and tau pathology serve as key roles in AD pathogenesis. At present, numerous pre-clinical studies or clinical trials focus on inducing or enhancing autophagy in AD treatments[114–118]. Activation of autophagy reduces Aβ and tau accumulation in AD animal models, which alleviates AD-related impairment in brains. Major regulations targeting different molecules lead to AMPK activation, mTORC1 inhibition, and transcription factor EB (TFEB) activation, which directly or indirectly enhance autophagy[19]. Therefore,we highlight and summarize the potential compounds and drugs that potentially play neuroprotective roles by enhancing autophagyin vivoor in clinical trials inTable 1andTable 2.
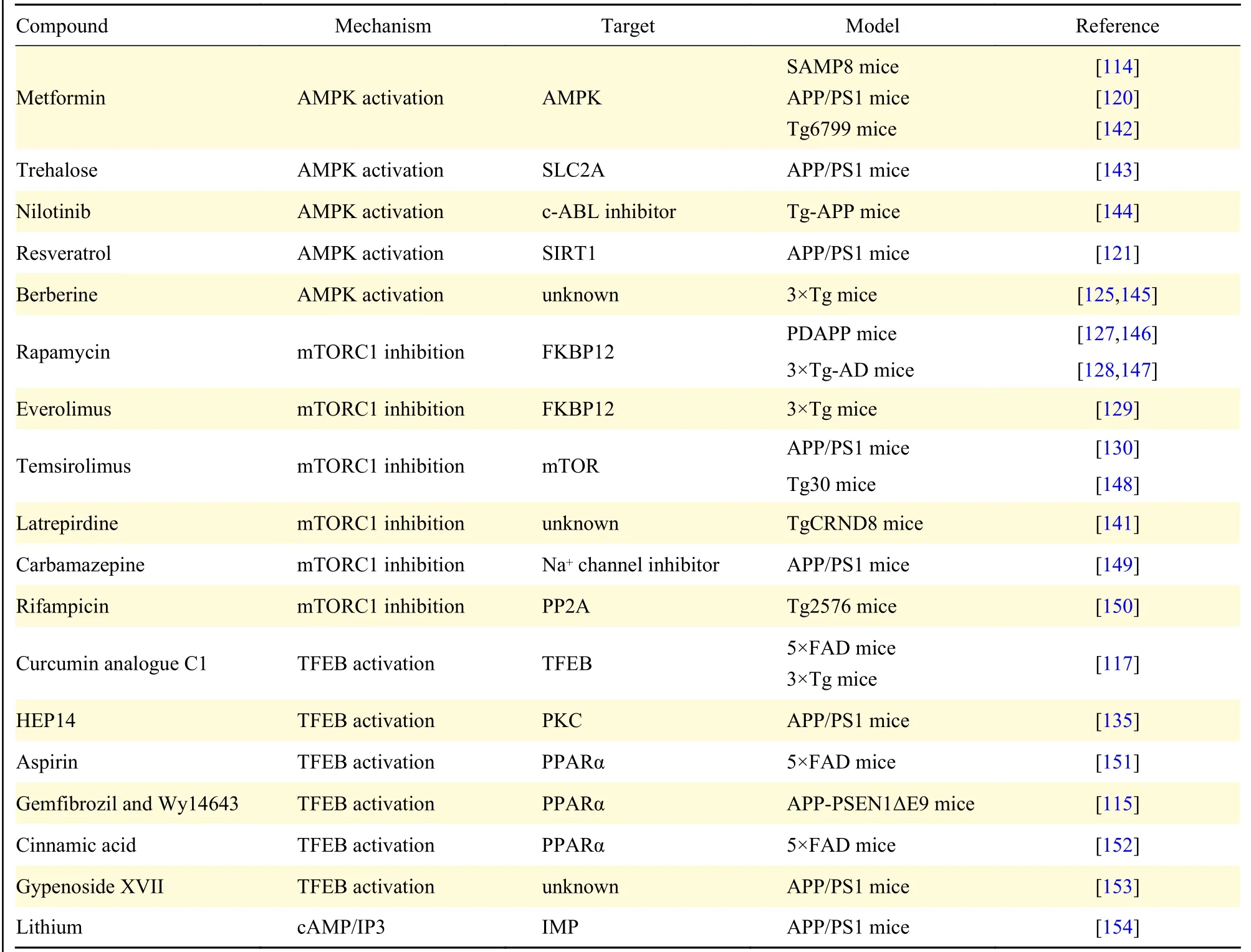
Table 1 Autophagy enhancers in Alzheimer's disease animal models
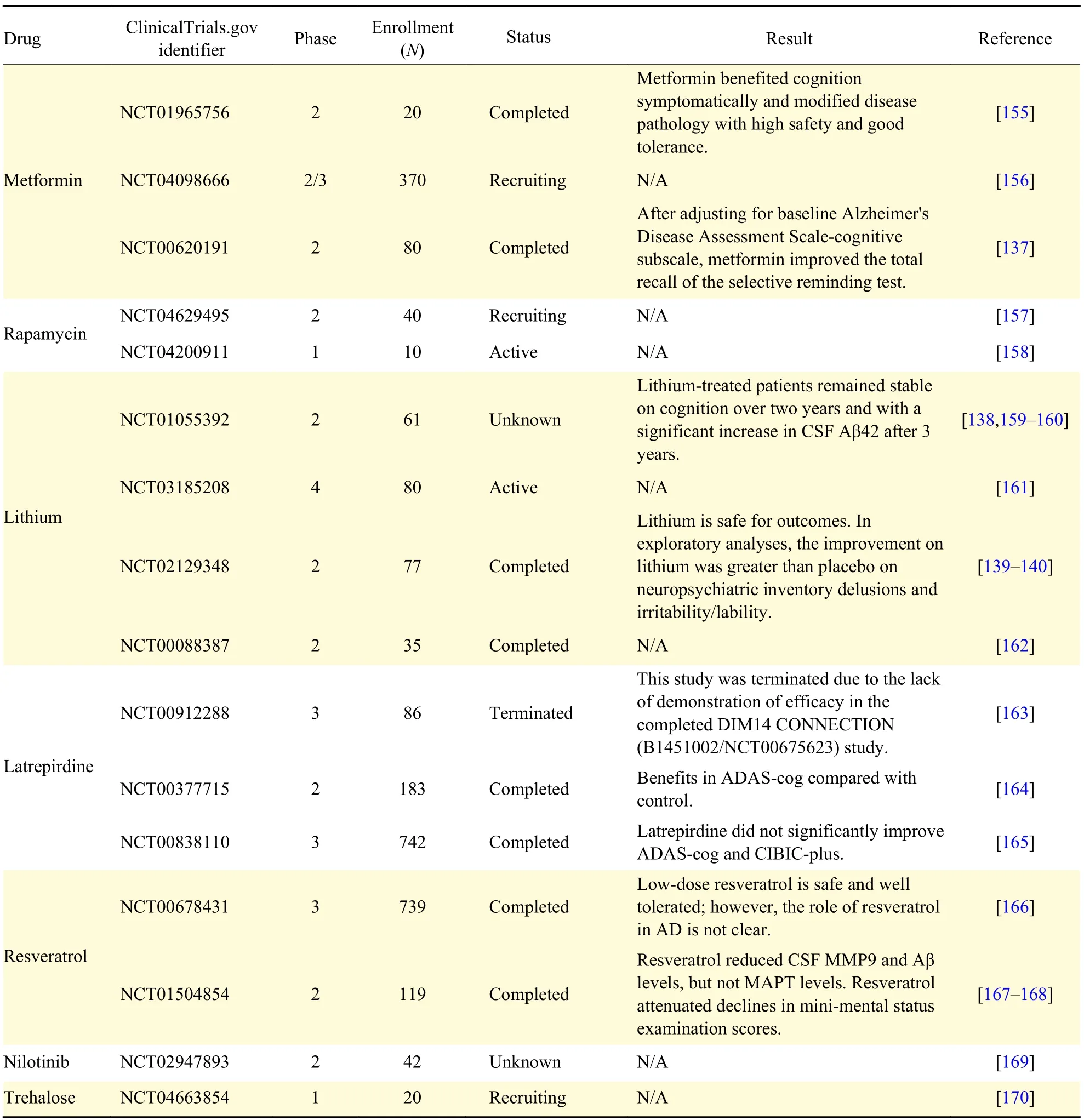
Table 2 Autophagy enhancers in clinical trials of Alzheimer's disease
Autophagy enhancers in pre-clinical animal models
Various small molecules, such as metformin,resveratrol, nilotinib and berberine, activate AMPK to induce autophagy, which plays neuroprotective roles in AD animal models[119]. Metformin is an antidiabetic drug that can activate AMPK, and improve cognitive dysfunctionviareducing Aβ plaque loading and phosphorylated tau levels in AD models[114,120];however, the role of Metformin in AD is controversial due to the lack of in-depth studies. Resveratrol, as a natural polyphenol widely distributed in edible food,can activate AMPK and bind to SIRT1 to induce autophagy. Autophagy-dependent and -independent effects of resveratrol on the Aβ metabolism were successively reported to explore the precise mechanism of resveratrol in AD[121–122]. Berberine is an AMPK activator that has multiple biological activities,including metabolic anti-diabetes and antihypercholesterolemia[123–124]. Berberine has been found to induce autophagy and improve learning and memory function by promoting Aβ degradation in AD animal models[125]. These findings imply that multiple AMPK-dependent autophagy enhancers exert neuroprotective effects in AD animal models.
Apart from AMPK activators, inhibiting mTOR is another important way to induce autophagy.Rapamycin, as a classical inhibitor of mTORC1,directly induces autophagy combatingneurodegenerative disorders, such as AD. Rapamycin can promote autophagy through binding to the cytosolic protein FKBP1A/FKBP12 (FK-binding protein 12)[126]. Furthermore, reduced levels of Aβ and rescued cognitive decline were detected after rapamycin treatment in 3XTg and hAPP(J20)mice[127–128]. However, the role of rapamycin in AD may be partly involved in other pathways, since rapamycin is a non-autophagy-specific compound.Given the side effects, such as glucose intolerance and hyperlipidemia caused by chronic rapamycin treatment, specific mTORC1 inhibitors, including Everolimus and Temsirolimus, have been developed.Everolimus has been found to inhibit autophagy,reduce Aβ levels and ameliorate cognitive deficits in AD mice[129]. In addition, inhibited autophagy and reduced levels of Aβ were widely found in the brain of APP/PS1 mice treated with Temsirolimus[130]. Other compounds, such as latrepirdine, carbamazepine and rifampicin, may be also promising anti-AD candidates. However, future studies on the underlying mechanisms of these compounds in AD are highly anticipated.
TFEB is an important transcription factor regulating cell function. Activated TFEB induces the expression of multiple autophagy-related genes[131], which promotes Aβ degradation and attenuates AD development through the autophagy-lysosomal pathway[132–133]. As previously mentioned, the autophagy-lysosomal pathway is impaired during AD progression. Therefore, upregulation of TFEB may a promising therapeutic strategy for AD targeting Aβ degradation through enhancing autophagy-lysosomal pathway[133]. Curcumin analogue C1 has also been shown to activate TFEB-mediated autophagylysosomal biogenesis, and further promote Aβ clearance in 5XFAD and 3XTg mice[117,134]. In addition to C1, HEP14 (5β-O-angelate-20-deoxyingenol) has been found to promote TFEB activation through binding to and activating PKCα and PKCδ[135]. This HEP14-mediated TFEB activation in the brain of APP/PS1 mice enhances the clearance of Aβ in these mice[135]. Other small molecule TFEB activators, including aspirin, cinnamic acid,gemfibrozil and gypenoside XVII, were associated with a decreased Aβ pathological changes in AD animal modelling. Overall, these findings provide a broad evidence for an emerging AD treatment strategy.
Autophagy enhancers in clinical trials of Alzheimer's disease
To date, multiple autophagy activators have been presented in clinical trials to test for their efficacy in AD patients. Current studies have reported that Metformin is safe and well-tolerated, improving cognitive deficiencies in AD patients[136–137].Additionally, a relatively small phase Ⅱ/Ⅲ trial including 370 patients has been tested to investigate the effects of Metformin on AD progression(NCT04098666). Although, rapamycin has been widely reported in autophagy activation and neuroprotection under various AD animal models, an investigation on its safety and feasibility for AD patients has only just begun (NCT04629495). By contrast, lithium has been well-studied through clinical AD trials. MCI patients who were treated with lithium for two years did not seem to have a significant cognitive decline according to the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) and the Clinical Dementia Rating scale (CDR-SoB) scores[138]. Additionally, a significant increase in CSF Aβ contents was detected in the lithium treatment group[138], which suggests that lithium has a neuroprotective role in AD patients.
Similarly, positive effects of lithium on behaviours of the AD patients have been demonstrated in recent studies[139–140]. Although latrepirdine had been found to alleviate neuropathic Aβviarestoring autophagy impairmentin vivo[141], clinical trials showed that this drug did not improve cognitive dysfunction, and two ongoing studies were terminated (NCT00912288 and NCT00838110). These differences in efficacy between animal models and AD patients may infer that latrepirdine has other functions unrelated to autophagy. However, other drugs, such as resveratrol,nilotinib and trehalose, have provided anti-AD effects according to different results. Collectively, clinical trials suggest that autophagy enhancers may be emerging therapeutic strategies for AD patients.However, given that autophagy enhancers have a wide variety of targets, large-scale studies of autophagic markers are still required.
Conclusions and perspectives
Autophagy is a necessary process involved in removing proteins prone to aggregation and neuronal intracytoplasmic deposition, both of which result in neurodegenerative disease. Autophagy is closely related to AD pathogenesis, and defective autophagy,which includes the accumulation of autophagic vesicles, decreased autophagic flux, failure of autophagosome maturation and defective autolysosome formation, which have been observed in AD. Impaired autophagy increases levels of Aβ production and reduces Aβ clearance, also it causes degradation failures in BACE1 and APP and activates the γ-secretase to promote Aβ production and accumulation. Correlation between defective autophagy and Aβ clearance failure has now been confirmed. Therefore, targeting autophagic regulation may provide a viable new therapeutic approach for treating AD. The findings of multiple studies suggest various potential therapeutic targets for autophagy, but their individual efficacies are yet to be investigated.
Fundings
This work was supported by the Construction Project of Capacity Improvement Plan for Chongqing Municipal Health Commission affiliated unit(2019NLTS001)-ZS03174, operating grant to Chongqing Key Laboratory of Neurodegenerative Diseases (Grant No. 1000013), Chongqing Talent Project [2000062], Overseas Students entrepreneurial fund (Grant No. 2000079), and Plan for High-level Talent Introduction (Grant No. 2000055).
Acknowledgments
The content in this article is the sole responsibility of the authors.
 THE JOURNAL OF BIOMEDICAL RESEARCH2023年1期
THE JOURNAL OF BIOMEDICAL RESEARCH2023年1期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Acknowledgments
- Washed microbiota transplantation stopped the deterioration of amyotrophic lateral sclerosis: The first case report and narrative review
- Peripheral CD4+CD8+ double positive T cells: A potential marker to evaluate renal impairment susceptibility during systemic lupus erythematosus
- Glioblastoma multiforme: Diagnosis, treatment, and invasion
- Acquired immunity and Alzheimer's disease
- Role of mitophagy in the hallmarks of aging