
Nuclear dissociation after the O 1s →(4Σ?u)3sσ excitation in O2 molecules
2022-08-31 09:55BochengDing丁伯承RuichangWu吳睿昌YunfeiFeng封云飛andXiaojingLiu劉小井
Chinese Physics B 2022年8期
關(guān)鍵詞:小井
Bocheng Ding(丁伯承) Ruichang Wu(吳睿昌) Yunfei Feng(封云飛) and Xiaojing Liu(劉小井)
1School Physical Science and Technology,ShanghaiTech University,Shanghai 201210,China
2Center for Transformative Science,ShanghaiTech University,Shanghai 201210,China
3University of Chinese Academy of Sciences,Beijing 100049,China
Keywords: nuclear dissociation,resonant excitation,spectator Auger
1. Introduction
With the x-ray photon energy below the O 1s ionization threshold, the O2molecule can be promoted to an excited state, either bound or dissociative, triggering relaxation processes such as soft x-ray photon emission, resonant Auger electron emission, and deformation of molecular structure.If the excited state is bound, the molecule will mainly decay by emitting an electron, while the excited electron may participate in Auger decay or standby like a spectator.[1,2]If the excited state is highly dissociative, competition between molecular motion and electron emission will occur. In the extreme case, Auger decay can even happen after molecular dissociation.[3–7]As discussed here in the context of the O2molecules, Feifelet al.[8]investigated the participator Auger decay after O 1s→Rydberg excited O2with the photon energies across theK-shell resonance region.Tanakaet al.[9]comprehensively measured a series of resonant spectator Augerelectron spectra of O2.
When an electron is excited to a Rydberg orbital, spectator Auger decay dominates the electronic decay processes and leaves the molecule to a two-hole-one-electron cationic state with two valence holes and one electron in a Rydberg orbital.[9,10]Because the Rydberg electron provides only partial screening of the repulsive force from the dicationic core,the two-hole-one-electron states are often dissociative.[11]Hikosakaet al.[12]found that most of the inner-valance-hole states of O+2are strongly dissociative,and end up with a neutral atom and a singly charged ion. A rather comprehensive calculation of the potential energy curves was reported long time ago.[13]Using Doppler-free kinetic release spectroscopy and multireference configuration interaction calculations, the potential energy curves of O2+2 were reported by Lundqvistet al.[14]
Compared with the singly and doubly ionized states, the dissociation of two-hole-one-electron states was much less investigated. Because either from an experimental point of view or from a theoretical point of view, the study of the twohole-one-electron state is very difficult.[11]In our recent work,we measured the correlation between the Auger electrons and ions using a high-resolution energy-resolved coincidence experimental setup, after O 1sσ →(4Σ?u)4pσexcitation in O2molecules at the photon energy of 541.8 eV.We described that the O+2two-hole-one-electron states dissociate into O+and O with a Rydberg electron, the results highlight how the spectator Rydberg electron participates in the dissociation of the Auger final states.[15]We also found that some weak channels correspond to dissociated O+and O without a Rydberg electron after time-consuming fine-tuning of parameters in data analysis, which points toward selectivity on the orbital size in the dissociation processes of the spectator Auger final states.[11]In this paper,we switch the measurement and analysis at the photon energy of 538.95 eV,which corresponds to O 1sσ →(4Σ?u)3sσcore excitation in O2.
2. Experimental method
Since the experimental method has already been described in detail in Refs. [11,15], only a brief description is given here. The experiments were carried out using the EPICEA setup at the PLEIADES beamline[16]at synchrotron SOLEIL in Saint-Aubin, France. This setup is specially designed for high kinetic-energy electron-ion coincidence measurement,which is composed of a double-toroidal electron analyzer(DTA)[17,18]and an ion TOF spectrometer.
The DTA selected the electron with an emission direction in the polar angle of 54±3?with respect to its symmetry axis and the azimuthal angles from 0?to 360?. The photon energy was set as 538.95 eV, which corresponds to O 1sσ →(4Σ?u)3sσcore excitation in O2,[9]and the light polarization was parallel to the symmetry axis of the setup. The DTA was operated with the pass energy of 80 eV, the central kinetic energy was set at 509 eV,and the electron energy resolution was about 0.7 eV.Opposite to the DTA,there is an ion TOF spectrometer incorporating 3D focusing mode. A pulsed±100 V ion extraction field (15 mm spacing) was triggered upon the detection of an Auger electron and pushed the ions toward the ion detector.The mass resolution of the nearly zero kinetic energy ions is about 1000, the ion momentum resolution is about 1.2–1.6 a.u., and the ion energy resolution was~0.3 eV.A random signal generator was used to simulate the false coincident event, and their contribution was subtracted according to the method described by Pr¨umperet al.[19]
3. Result and discussion
The resonant Auger electron energy spectrum following O 1sσ →(4Σ?u)3sσexcitation in O2at the photon energy of 538.95 eV is shown in Fig. 1, which covers the kineticenergy range from 504 eV to 514 eV(corresponding to a binding energy range from 24.95 eV to 34.95 eV). Peaks above 507 eV are much broader than the peaks below. Because the former peaks come from O2(1s?1σ?) state, whose potential energy curve is highly dissociative and covers a broad energy range within the Franck–Condon region.[4]In contrast,the later peaks come from O2(1s?14Σ?u)3sσbound state, so widths of these peaks are much narrower. To our knowledge,the assignments of the former peaks have not been reported in literature,while the assignments of the latter are adopted from Tanakaet al.[9]
The ion time-of-flight spectrum is shown in Fig. 2. The peak of O+2is very narrow because its kinetic energy can only be contributed by the thermal motion and the recoil during electron emission. The profile of O+is very broad and splits into two wings,because O 1sσ →(4Σ?u)3sσRydberg excitation is sitting on the top of O 1s→σ?resonance,[6]which occurs preferentially when the molecular axis is parallel to the polarization vector of the incoming photons and the axis of TOF spectrometer,and the molecular orientation is preserved during molecular fragmentation,which is generally faster than molecular rotation in this case. We cannot present here the electron spectrum related to O+2due to very low statistics.
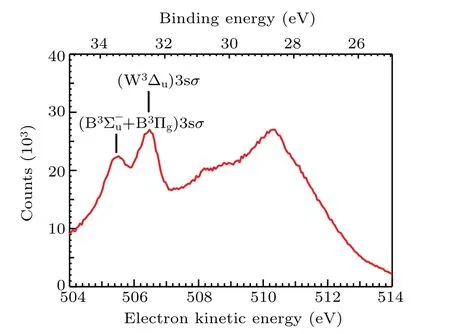
Fig.1. The resonant Auger electron spectrum after O 1sσ →(4Σ?u)3sσ excitation in O2 at the photon energy of 538.95 eV.Assignments of two spectator Auger final states are adopted from Tanaka et al.[9]
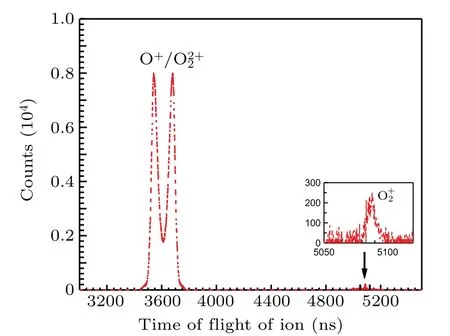
Fig.2. Ion time-of-flight spectrum recorded after O 1sσ →(4Σ?u)3sσ excitation in O2 at the photon energy of 538.95 eV. The inset shows a magnified O2+ peak.

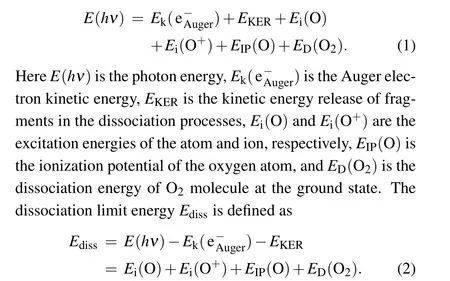
The dissociation limits energiesEdissare calculated using Eq.(1)and assigned using Eq.(2)with the energy values from the NIST atomic database,[20]as listed in Table 1. The variousEdiss’s are represented by the red dashed tilted lines in Fig.3.Here,we label them as I,II,III,and IV.In Table 1,we also list the other dissociation limits which do not show observable intensities, as indicated by circles in Table 1 and by four green dashed tilted lines in Fig.3.
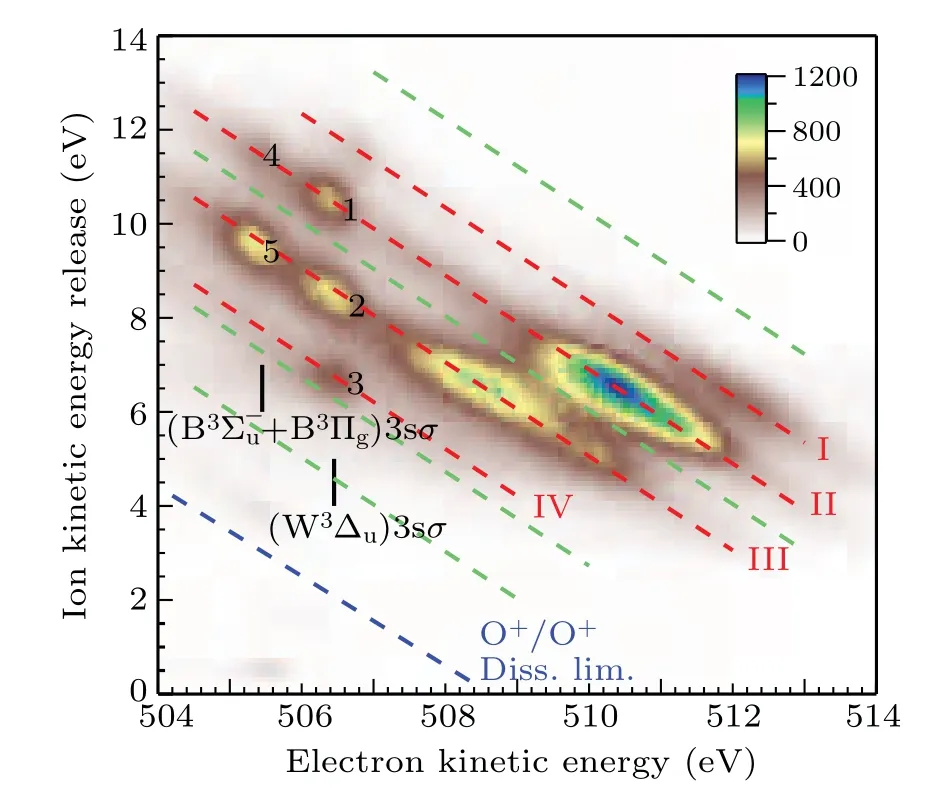
Fig.3. The correlation between the kinetic energy release of the fragments and the kinetic energy of the Auger electron at the photon energy of 538.95 eV.The red dashed tilted lines I,II,III,and IV represent groups of dissociation limits after spectator Auger decay. The green dashed tilted lines indicate the missing dissociate limits. The blue dashed tilted line indicates the appearance threshold of O+ +O+. The assignments of spectator Auger lines are adopted from Tanaka et al.[9]

Table 1. Assignment of the dissociation limit energy Ediss for the features observed in Fig.3. Symbols ○are used to mark missing features in Fig.3. The unit of energy is eV.
As indicated by green dashed tilted lines in Fig.3,it can be seen that the first,fourth,seventh,and eighth limits do not show observable intensities. This puzzle can be explained by examining a comprehensive summary of potential curves of O+2calculated by Beedeet al.[13]Among them, we choose repulsive potential curves with proper symmetry,correct binding energy in the Franck–Condon region, and dissociate into the limits given in Table 1. We find that the2,4?u, the2,4Σ?u,and the2,4Πgfrom Beedeet al.[13]are the candidates. As a result,only the second,third,fifth,and sixth limits should exist,which agrees reasonably well with the observed I, II, III, and IV limits in Table 1.

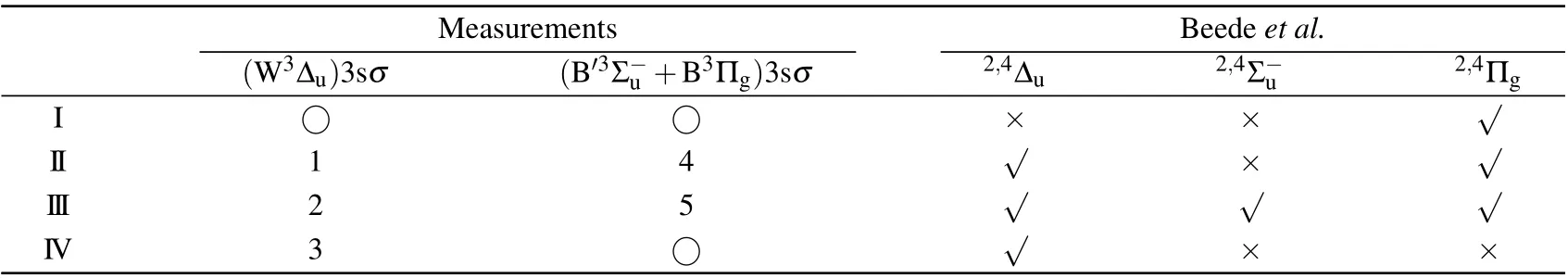
Table 2. Assignment of the dissociation channels after O 1sσ →(4Σ?u)3sσ excitation in O2. Number and ○symbols are used to mark observable and missing features in our measurements,respectively.Based on the potential curves reported by Beede et al.,√and×symbols are used to indicate the feature should exist and not,respectively.
4. Conclusions

Acknowledgements
Project supported by the National Natural Science Foundation of China (Grant No. 11574020), the Project of Thousand Youth Talents in China,and the Starting Grant of ShanghaiTech University.
猜你喜歡
發(fā)明與創(chuàng)新(2022年30期)2022-10-03
現(xiàn)代家長(zhǎng)(2020年3期)2020-04-20
莫愁·小作家(2019年10期)2019-09-10
老友(2019年5期)2019-05-26
中學(xué)生天地(B版)(2018年12期)2018-12-22
方圓(2018年4期)2018-03-09
出版人(2015年8期)2015-09-09
幸福家庭(2014年6期)2014-09-10
語(yǔ)文教學(xué)與研究(讀寫(xiě)天地)(2012年10期)2012-05-10

- Chinese Physics B的其它文章
- Direct measurement of two-qubit phononic entangled states via optomechanical interactions
- Inertial focusing and rotating characteristics of elliptical and rectangular particle pairs in channel flow
- Achieving ultracold Bose–Fermi mixture of 87Rb and 40K with dual dark magnetic-optical-trap
- New experimental measurement of natSe(n,γ)cross section between 1 eV to 1 keV at the CSNS Back-n facility
- Oscillation properties of matter–wave bright solitons in harmonic potentials
- Synchronously scrambled diffuse image encryption method based on a new cosine chaotic map