
Genomic landscape of T-cell lymphoblastic lymphoma
2022-05-23 07:36:14ZhaomingLiYueSongMingzhiZhangYimingWeiHangRuan
Zhaoming Li ,Yue Song ,Mingzhi Zhang ,Yiming Wei,2 ,Hang Ruan,2
1Department of Oncology,the First Affiliated Hospital of Zhengzhou University,Zhengzhou 450052,China;2State Key Laboratory of Esophageal Cancer Prevention &Treatment and Henan Key Laboratory for Esophageal Cancer Research,the First Affiliated Hospital of Zhengzhou University,Zhengzhou 450052,China
Abstract Objective:T-cell lymphoblastic lymphoma (T-LBL) is an aggressive neoplasm of precursor T cells,however,detailed genome-wide sequencing of large T-LBL cohorts has not been performed due to its rarity.The purpose of this study was to identify putative driver genes in T-LBL.Methods:To gain insight into the genetic mechanisms of T-LBL development,we performed whole-exome sequencing on 41 paired tumor-normal DNA samples from patients with T-LBL.Results:We identified 32 putative driver genes using whole-exome sequencing in 41 T-LBL cases,many of which have not previously been described in T-LBL,such as Janus kinase 3 (JAK3),Janus kinase 1 (JAK1),Runtrelated transcription factor 1 (RUNX1) and Wilms’ tumor suppressor gene 1 (WT1).When comparing the genetic alterations of T-LBL to T-cell acute lymphoblastic leukemia (T-ALL),we found that JAK-STAT and RAS pathway mutations were predominantly observed in T-LBL (58.5% and 34.1%,respectively),whereas Notch and cell cycle signaling pathways mutations were more prevalent in T-ALL.Notably,besides notch receptor 1(NOTCH1),mutational status of plant homeodomain (PHD)-like finger protein 6 (PHF6) was identified as another independent factor for good prognosis.Of utmost interest is that co-existence of PHF6 and NOTCH1 mutation status might provide an alternative for early therapeutic stratification in T-LBL.Conclusions:Together,our findings will not only provide new insights into the molecular and genetic mechanisms of T-LBL,but also have tangible implications for clinical practice.
Keywords:T-cell lymphoblastic lymphoma;PHF6;NOTCH1;mutation
Introduction
T-cell lymphoblastic lymphoma (T-LBL) is a rare and aggressive neoplasm of precursor T cells,with an incidence less than 2% of all non-Hodgkin’s lymphomas (NHL) and a bimodal age distribution,with higher frequencies in individuals younger than 20 years and in those older than 50 years.The incidence is higher in children,where TLBL represents the second most common subtype of NHL(1). Because T-LBL shares common clinical,morphological and immunophenotypic features with T-cell acute lymphoblastic leukemia (T-ALL),they have often been thought to represent a spectrum of a single disease,differing by the extent of bone marrow infiltration (2).However,there is still ongoing discussion on whether TLBL and T-ALL are two distinct entities (3).Currently,pathogenetic and molecular biological knowledge of TLBL is extremely limited,partly due to the rarity of this disorder and the difficulty in obtaining appropriate material for research.Therefore,it remains elusive whether there might be additional differences in molecular genetics which distinguish T-LBL from T-ALL.
Previous candidate gene mutational and cytogenetic analyses have identified recurring genetic alterations in TLBL patients (4,5),for example,NOTCH1and F-Box and WD Repeat Domain Containing 7 (FBXW7) mutations were found in 55% of T-LBL patients,and associated with better prognosis (6).Genes of the PI3K-AKT signaling pathway were commonly mutated in T-LBL (7).Although these studies have provided important insights into the oncogenic pathways that drive lymphomagenesis in TLBL,there are relatively few data from unbiased,genomewide sequencing approaches,except one study with a relatively small cohort of patients (8).
Here we carried out whole-exome sequencing in a cohort of 41 individuals with T-LBL to illustrate the spectrum and constellations of genetic alterations in T-LBL,and identify molecular genetic markers for risk stratification of these patients.
Materials and methods
Samples
In total,41 formalin-fixed,paraffin-embedded (FFPE)tumor tissues from T-LBL patients were obtained from the First Affiliated Hospital of Zhengzhou University.All cases were reviewed and interpreted independently by three experienced pathologists,and the diagnoses were made according to the current World Health Organization classification criteria.The diagnosis was confirmed by two pathologists.The study was conducted in accordance with the Declaration of Helsinki and with approval of the Institutional Review Board of the First Affiliated Hospital of Zhengzhou University.Signed informed consent was obtained from all patients.
Whole-exome sequencing,mutation calling and annotation
To identify somatic genomic variants associated with TLBL,we performed whole-exome sequencing on 41 tumor samples and their matched normal tissue (whole-blood samples or skin) (Novogene Bioinformatics Institute,Beijing,China).The whole-exome sequencing,mutation calling and annotation were performed as previously described (9).
Sanger validation of mutation calls
We performed PCR amplification and Sanger sequencing on a total of 115 putative somatic single nucleotide variants(SNVs) and insertions and deletions (InDels) of the top mutated genes [NOTCH1,PHF6,JAK3,JAK1,NRAS,ETS variant transcription factor 6 (ETV6),FBXW7,RUNX1,SUZ12andWT1] identified by next-generation sequencing were verified by Sanger sequencing.Excluding the loci that failed in PCR,94 of 101 SNVs and InDels (93%) were successfully validated.
Data availability
Data supporting the main findings of this study are available within the article and its supplementary information and supplementary data files.Whole-exome sequencing data were deposited into the NCBI Sequence Read Archive with the BioProject ID:PRJNA473585 and Title:T-cell lymphoma sequencing.
Statistical analysis
Results
Whole-exome sequencing of T-LBL samples
To gain insight into the genetic mechanisms of T-LBL development,we performed whole-exome sequencing on 41 paired tumor-normal DNA samples from patients with T-LBL.In the entire cohort,most of patients were adults(>18 years:75.6%) with a median age of 26 (range,11?60)years,73.2% were males (male/female:2.7),and 80.5%presented with an advanced stage (stage IV).Clinical characteristics of the patient cohort are summarized inSupplementary Table S1.The mean sequencing depth was 217×,and a mean of 96.6% of the target sequence was covered to a depth of at least 50× after excluding the duplicates.A total of 1,599 non-silent mutations (median,33;range,4?124) were identified (Figure 1A).
To identify whether mutagenic processes are operative in T-LBL,we analyzed the mutation spectrum in a total of 41 T-LBL patients.The predominant type of substitution was C to T transition at NpCpG sites in T-LBL (Figure 1B).Combined non-negative matrix factorization clustering and correlation with the 30 curated mutational signatures defined by the Catalog of Somatic Mutations in Cancer(COSMIC) database (10) revealed three predominant signatures in T-LBL (Figure 1C).The predominant matched signature was Signature 1 (cosine similarities,0.94;Supplementary Figure S1),which was found in all tumor types and was thought to result from age-associated accumulation of 5-methylcytosine deamination events.
We further evaluated the relationship between the somatic non-silent mutation burden and clinical features of T-LBL.The results showed that somatic non-silent mutation burden was not associated with gender,stage,lactase dehydrogenase,etc.Furthermore,survival analysis revealed no significant correlation between somatic nonsilent mutation burden and OS of T-LBL patients (P=0.26;Figure 1D).The median tumor burden was applied as the cut-off value.
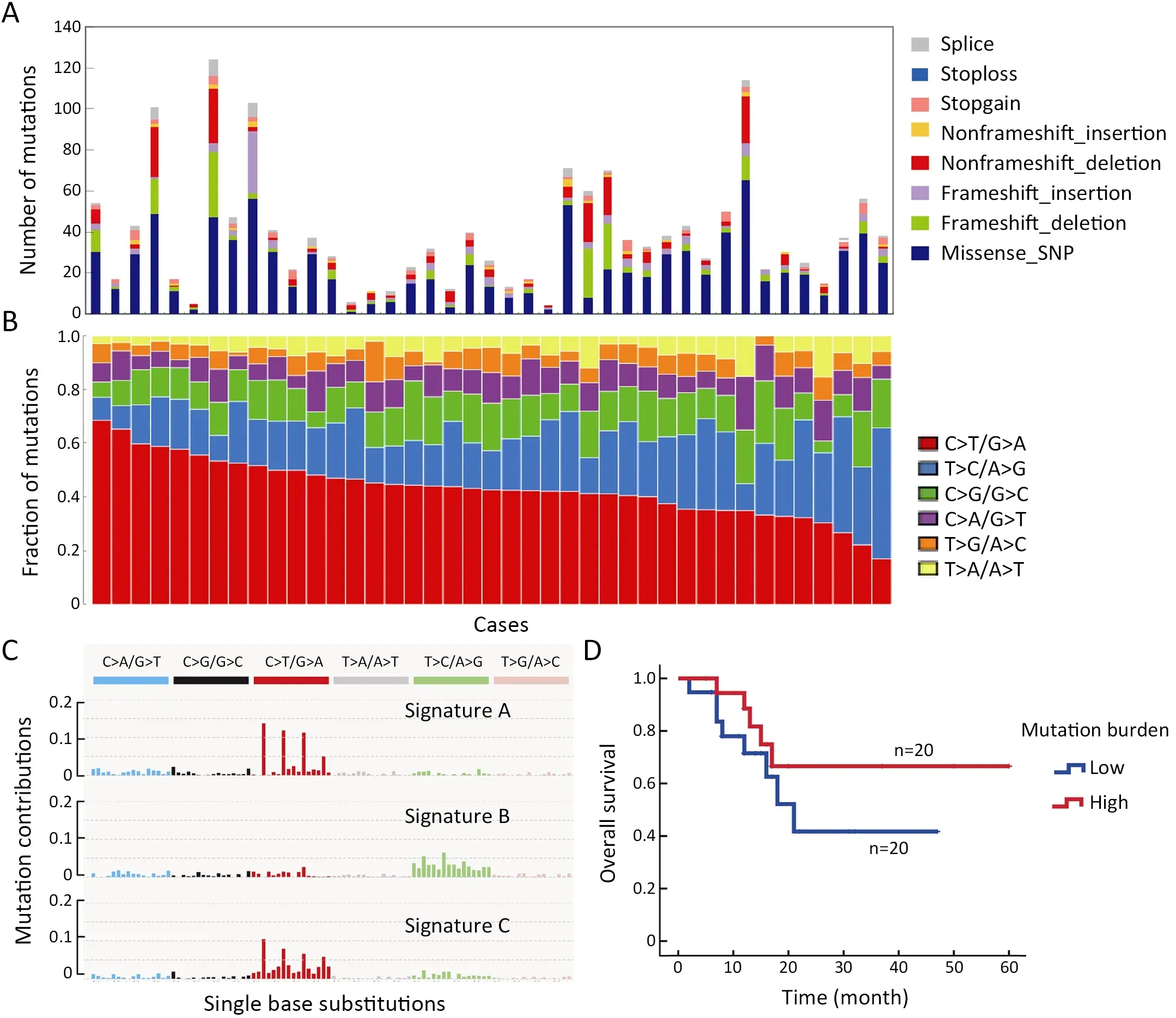
Figure 1 Whole-exome sequencing of 41 cases of T-LBL.(A) Number and type of non-silent somatic mutations;(B) Mutation spectrum of T-LBL;(C) Three dominant signatures identified in T-LBL by combined non-negative matrix factorization clustering and correlation,with 30 curated mutational signatures defined by COSMIC database;and (D) Association of somatic non-silent mutation burden with overall survival in T-LBL (log rank P=0.26).T-LBL,T-cell lymphoblastic lymphoma;COSMIC,the Catalog of Somatic Mutations in Cancer.
Significant mutated genes
The mutational significance detection tools MuSiC (11)were used to identify potential driver mutations.Overall,we identified 32 genes with a false discovery rate (FDR) less than 0.1.Each of them affected at least two cases (Figure 2A,Supplementary Table S2.These included the NOTCH signaling genesNOTCH1(20;48.78%),FBXW7(6;14.63%)andNOTCH3(3;7.32%);JAK-STAT signaling genesJAK3(14;34.15%),JAK1(11;26.83%) andIL7R(4;9.76%);epigenetic modifiers includingPHF6(15;36.59%),SUZ12(5;12.20%),ASXL Transcriptional Regulator 1 (ASXL1) (4;9.76%),chromodomain helicase DNA binding protein 4(CHD4) (4;9.76%),CCCTC-binding factor (CTCF) (3;7.32%),enhancer of zeste homolog 2 (EZH2) (3;7.32%)and embryonic ectoderm development (EED) (2;4.88%);transcriptional regulatory genesETV6(7;17.07%),RUNX1(6;14.63%),WT1(5;12.20%),SPI1(2;4.88%) and GATAbinding protein 3 (GATA3) (2;4.88%);RNA-processing geneU2AF1(3; 7.33%); PI3K-AKT signaling gene phosphatase and tensin homolog (PTEN) (3;7.32%);RAS signaling geneNRAS(7;17.07%);and others.Mapping of mutation sites of these frequently mutated genes are shown inFigure 2BandSupplementary Figure S2.Most of them have been reported in T-ALL (12) except the following genes,CDC27(5; 12.20%),MTRNR2L2(4; 9.76%),COL6A5(4;9.76%) andTMEM200C(3;7.32%).To explore their pathogenic role in T-LBL,we clustered somatic nonsilent mutated genes according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) categories.Mutated genes were significantly enriched in categories of NOTCH,JAKSTAT,PI3K-AKT signaling pathway and transcriptional misregulation in cancer,indicating that abnormalities of these key signaling pathways may have substantial roles in T-LBL pathogenesis.
We also examined the tumor clonality in each individual by using PyClone (13).Subclonal mutations were frequently detected in many driver genes (Supplementary Figure S3A),suggesting that subclonal evolution is a hallmark of T-LBL,which is consistent in T-ALL (12).In this study,we found thatJAK3,NOTCH1andJAK1driver mutations were clonal events in 26.83% (11/41),12.19%(5/41) and 12.19% (5/41) cases,respectively.Notably,NOTCH1is the most frequently mutated gene,with 27 sequence mutations identified in 20 cases.Of the 27 mutations,5 (18.51%) were subclonal.JAK3is another most commonly mutated gene,with 18 sequence mutations identified in 14 cases.However,in contrast toNOTCH1,most of these mutations (15/18;83.33%) were clonal.These data suggested thatJAK3might play an important part in the initiation of T-LBL.
The mutation relation test (MRT) was used to reveal correlations and mutual-exclusion relationships among significantly mutated genes in T-LBL.Resultsshowed thatJAK3positively correlated withJAK1(P<0.01,Supplementary Figure S3B).There is also evidence of the mutual exclusion ofJAK3andETV6(P<0.01) orNRAS(P=0.04) mutations (Supplementary Figure S3B).It was therefore proposed that,for the cases with no driver mutation inJAK3,mutations inETV6orNRASmight represent an independent path to T-LBL.
Somatic copy number alterations (SCNAs)
We further examined SCNAs in T-LBL.SCNAs analysis using genomic identification of significant targets in cancer(GISTIC) identified 58 focal amplifications and 10 focal deletions (Figure 3,Supplementary Table S3).Inconsistent with previously reports (14),amplifications of 6q23.3(24.39%),9q34.3 (21.95%),3q29 (24.39%) and 21q22.3(31.71%) recurrently occurred in our analysis.Additionally,amplification of 14q11.2 (36.59%),7q22.1 (19.51%) and 10q26.3 (26.83%) were also commonly found.Focal amplifications targeted oncogenes includedNOTCH2(1q21.1),AHNAK2(14q32.33),MACF1(1p34.3),NFATC1(18q23),PRKCA(17q24.2),MYB(6q23.3),MUC4(3q29)andMALAT1(11q13.1).
In addition to previously reported genomic deletions of 7p14.1 (TCRγ,43.90%),4q35.2 (DUX4,36.59%) and 9p21.3 (CDKN2A,24.39%) (14),several novel recurrent focal deletions were also identified,including 17q11.2(31.71%),7q34 (29.27%),10q26.3 (26.83%),5q23.3(21.95%),12p12.3 (21.95%),11p13 (17.07%) and 9p23(14.63%).The known tumor suppressors targeted by these novel focal deletions were neurofibromatosis type 1 (NF1)(17q11.2),transcription factor 7 (TCF7) (5q23.3) andWT1(11p13),suggesting that these genes might play vital roles in the lymphomagenesis of T-LBL.
Genetic differences between T-LBL and T-ALL
Although T-LBL shows many features in common with TALL,there is still ongoing discussion on whether T-ALL and T-LBL are two separate entities (3).We therefore investigated whether T-LBL are genetically distinct from T-ALL.First,we compared the frequencies of the most commonly mutated genes in T-LBL with those in T-ALL.Mutation data of T-ALL were obtained from the recent published genome-wide sequencing of a large T-ALL cohort (n=264) (12).The mutational profiles of T-LBL and T-ALL are shown inFigure 4,Supplementary Figure S4.We found that mutations inPHF6(36.6%),JAK3(34.2%),JAK1(26.8%),ETV6(17.1%),RUNX1(14.6%) andSUZ12(12.2%) occurred significantly more frequently in T-LBL than in T-ALL (Supplementary Table S4;P<0.05;Chisquare or Fisher exact test).Moreover,mutations inCDC27(12.2%),MTRNR2L2(9.8%),COL6A5(9.8%),TMEM200C(7.3%),CATSPER4(4.9%) andKRT38(4.9%) occurred almost exclusively in T-LBL.On the other hand,the most frequent mutated genes in T-ALL such asFAT1,RPL10,SMARCA4,BCL11BandUSP7,were seldom found in TLBL cases.
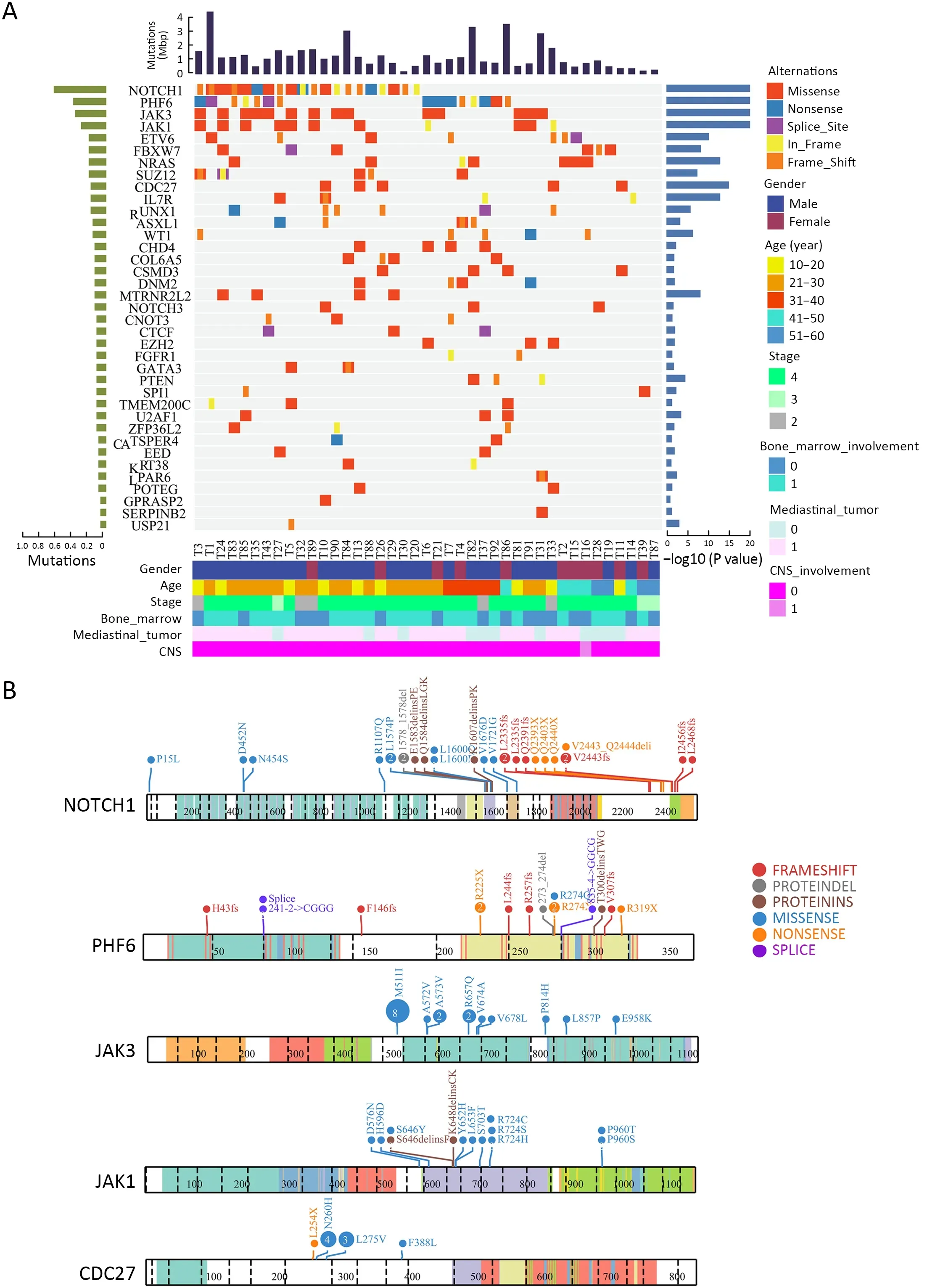
Figure 2 Significantly mutated genes identified by whole-exome sequencing in T-LBL.(A) Mutational significance was determined for SNV and InDels from the 41 sequenced cases using MuSiC.Significantly mutated genes are ranked according to mutation frequency.Samples are displayed as columns,and mutations are colored according to the type of alteration.The frequency and P value as determined by MuSiC are shown;(B) ProteinPaint visualizations of recurrently mutated genes in T-LBL.T-LBL,T-cell lymphoblastic lymphoma;SNV,single nucleotide variants;InDels,insertions and deletions.
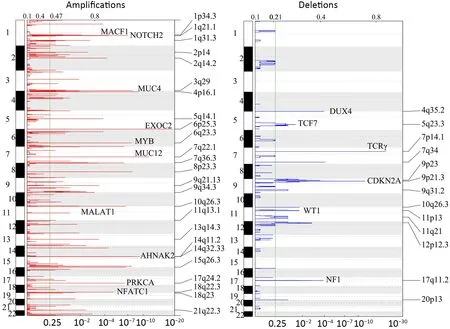
Figure 3 Copy number alterations in T-LBL.Global chromosomal amplifications (shown in red) and deletions (shown in blue) identified by whole-exome sequencing of 41 T-LBL cases;GISTIC analysis of cases reveals chromosomal regions that are significantly deleted/amplified.T-LBL,T-cell lymphoblastic lymphoma;GISTIC,genomic identification of significant targets in cancer.
Second,the most common SCNAs in T-LBL and TALL were compared in our study.As shown inFigure 3,focal amplifications of 1q44 and 6q23.3,and focal deletions of 7p14.1,11p13,17q11.2 and 9p21.3 were found in both T-LBL and T-ALL cases.The affected known oncogenes or tumor suppressor genes includedMYB,CDKN2A,NF1andWT1.Furthermore,the frequencies of genes targeted by these most common SCNAs were compared between TLBL and T-ALL (Figure 4A,Supplementary Figure S4).Deletion ofCDKN2A,a key negative regulator of cell cycle,was much more common in T-ALL than in T-LBL(P=0.0001).On the contrary,deletions of tumor suppressors such asNF1(P<0.001),WT1(P<0.001),SUZ12(P=0.004) andTCF7(P=0.022) were found to occur predominantly in T-LBL cases (Table 1).
Third,integrated analysis of sequence mutation and copy-number alteration data identified five functional pathways that were recurrently mutated in T-LBL,including JAK-STAT signaling (58.5%),NOTCH signaling (56.1%),cell cycle signaling (46.3%),PI3K-AKT signaling (17.0%) and RAS signaling (34.1%) (Figure 4B,Table 2).As compared to T-ALL,mutations in JAK-STAT signaling (T-LBLvs.T-ALL,58.5%vs.24.6%,respectively;P<0.001) and RAS signaling (T-LBLvs.TALL,34.1%vs.13.6%,respectively;P=0.003) were predominantly observed in T-LBL.In contrast,mutations in NOTCH and cell cycle signaling pathways were altered at higher frequencies in T-ALL than in T-LBL.Collectively,these data indicated that T-LBL might have a different profile of genetic alterations distinguishing it from T-ALL.
NOTCH1/PHF6 mutations predict favorable prognosis for patients with T-LBL
Currently,identification of biological markers that can predict outcome in T-LBL has been hampered by therarity of this disorder.In the present study,we investigated the association of genetic mutations with clinical outcome in 41 T-LBL cases.Genes with mutational frequency more than 10% were included in our analysis.The univariate analysis showed thatNOTCH1andPHF6mutations were significantly associated with clinical outcome(Supplementary Table S5).In consistent with previous reports,patients withNOTCH1mutations had improved OS (P=0.01) and PFS (P=0.002) compared with individuals without mutations (Figure 5A,B).Of note,PHF6mutations were also significantly correlated with good prognosis in TLBL patients.The estimated three-year OS rates of patients withPHF6mutations and those without mutations were (93.3±6.4)% and (41.1±11.5)%,respectively (P=0.01;Figure 5C).The estimated three-year PFS rates of the two groups of patients were (72.2±15.1)% and (28.7±10.4)%,respectively (P=0.004;Figure 5D).Moreover,a Cox multivariate regression model was conducted in our study.After controlling for clinical factors (age,stage and treatment),onlyNOTCH1orPHF6mutation status is independent prognostic markers for better outcomes in TLBL (Supplementary Table S5).
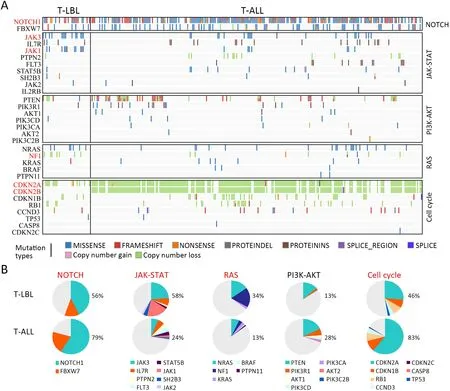
Figure 4 Genetic differences between T-LBL and T-ALL.(A) Frequency of gene mutations and CNVs between T-LBL (n=41) and TALL (n=264) were compared by χ2 test or Fisher’s exact test.P<0.05 is highlighted in red;(B) Key signaling pathways affected by frequent somatic mutations in T-LBL and T-ALL were compared.The majority of pathways showed significant associations between these two groups (P<0.05,χ2 test).T-LBL,T-cell lymphoblastic lymphoma;T-ALL,T-cell acute lymphoblastic leukemia;CNV,copy number variation.
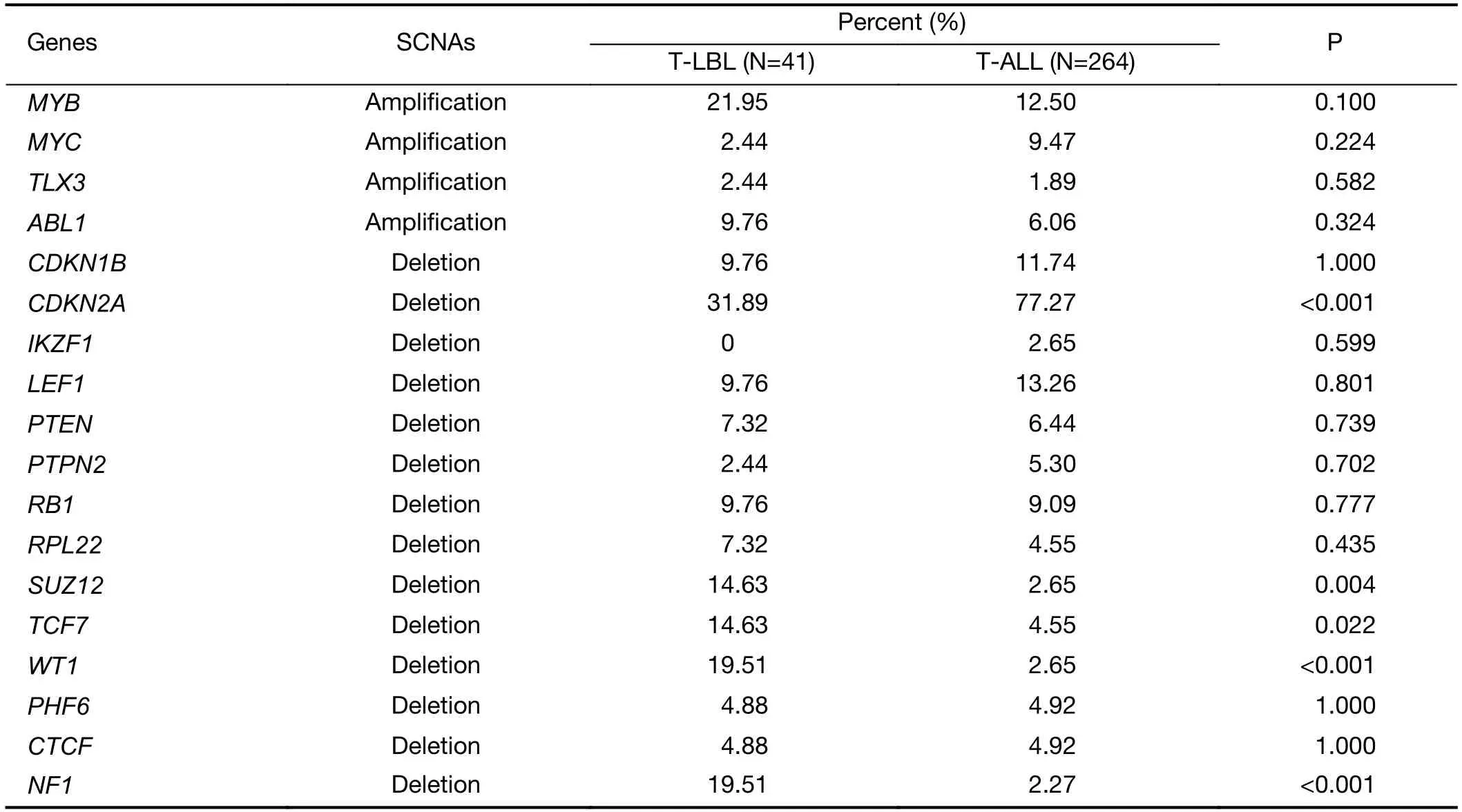
Table 1 Frequencies of genes targeted by these most common SCNAs in T-LBL and T-ALL
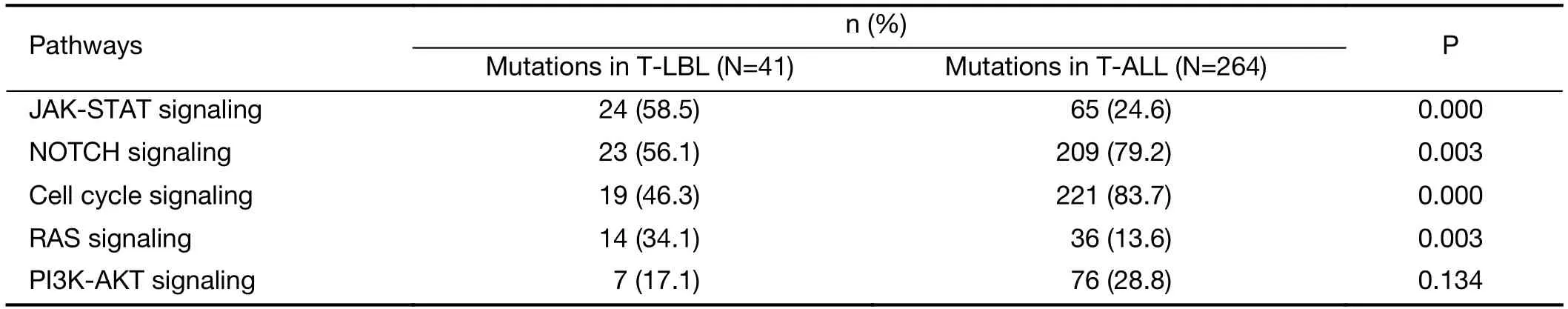
Table 2 Frequencies of recurrently mutated pathways in T-LBL and T-ALL
NOTCH1andPHF6were the two most frequently mutated genes in T-LBL,which affected 68.3% (28/41) of all patients in our study.After combining two genetic mutations,patients were divided into three groups according to the two gene mutation status:those with wildtypeNOTCH1and wild-typePHF6(n=13),those with mutations in eitherNOTCH1orPHF6(n=21),and those had bothNOTCH1andPHF6mutations (n=7).The result showed thatNOTCH1andPHF6wide-type patients showed an exceedingly poor prognosis,whereas patients with coexistence of the two mutations showed good response to current therapies and had an extremely good prognosis(OS,P=0.001;Figure 5E;PFS,P<0.001;Figure 5F).Accordingly,T-LBL patients in our study can be stratified into three groups with distinct prognoses:a low-risk subgroup (with co-existence of bothNOTCH1andPHF6mutations),an intermediate-risk subgroup (with mutations in eitherNOTCH1orPHF6) and a high-risk subgroup (with wild-typeNOTCH1andPHF6).Consequently,PHF6andNOTCH1might provide an alternative for therapeutic stratification in T-LBL.
Figure S1 Combined non-negative matrix factorization clustering and correlation with 30 curated mutational signatures defined by COSMIC database.COSMIC,the Catalog of Somatic Mutations in Cancer.

Figure S2 ProteinPaint visualizations of recurrently mutated genes in T-LBL.T-LBL,T-cell lymphoblastic lymphoma.
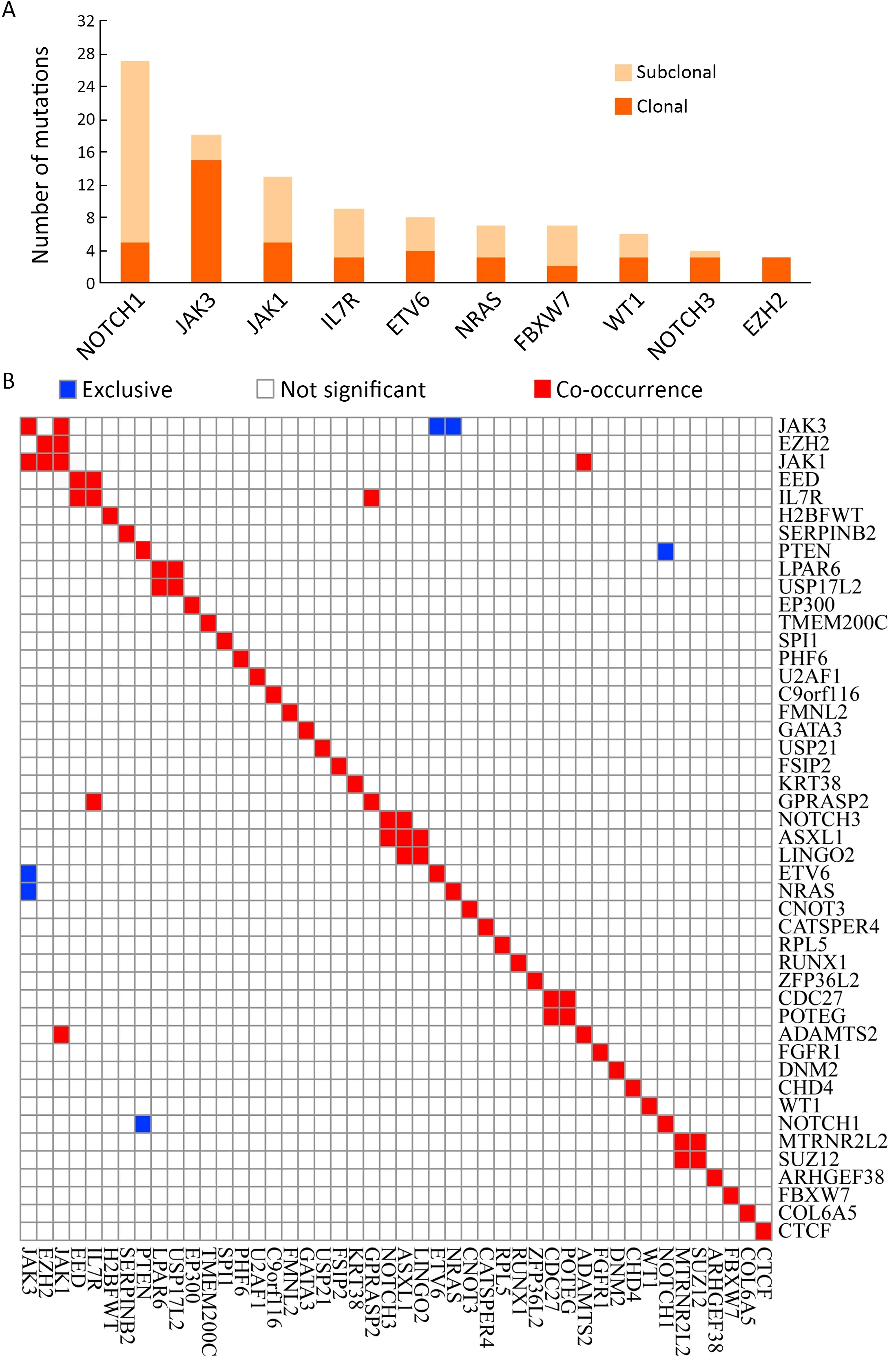
Figure S3 Tumor clonality analysis and correlation of genetic alterations.(A) Tumor clonality of each tumor was examined by using PyClone,and the numbers of subclonal and clonal gene mutations in frequently mutated genes are shown;(B) Hierarchical clustering using Ward’s minimum variance method of paired gene correlation analysis showed gene-based association between lesions in T-LBL.T-LBL,T-cell lymphoblastic lymphoma.
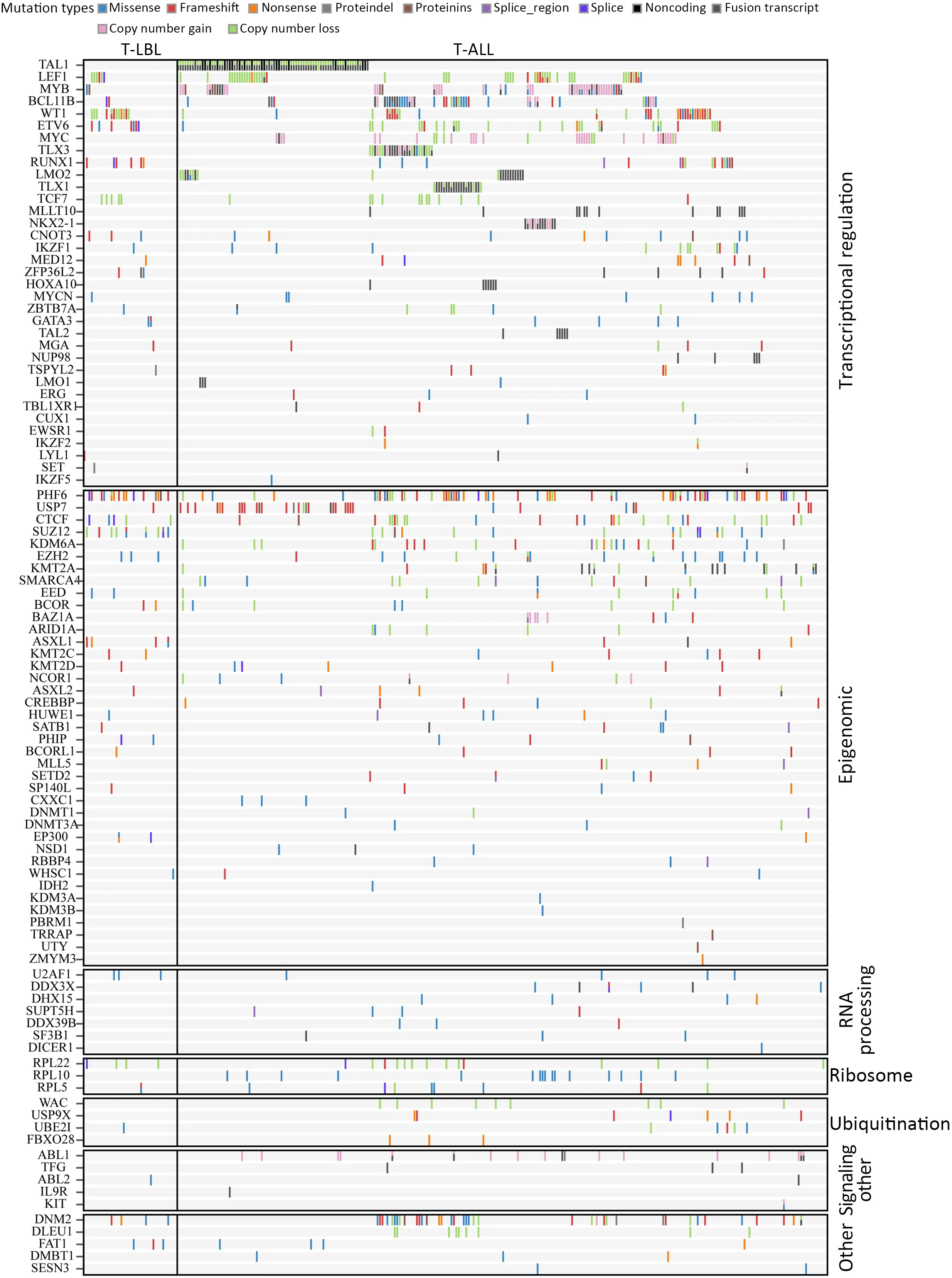
Figure S4 Frequency of gene mutations and CNVs between T-LBL (n=41) and T-ALL (n=264) were compared by χ2 test or Fisher’s exact test.CNV,copy number variation;T-LBL,T-cell lymphoblastic lymphoma;T-ALL,T-cell acute lymphoblastic leukemia.
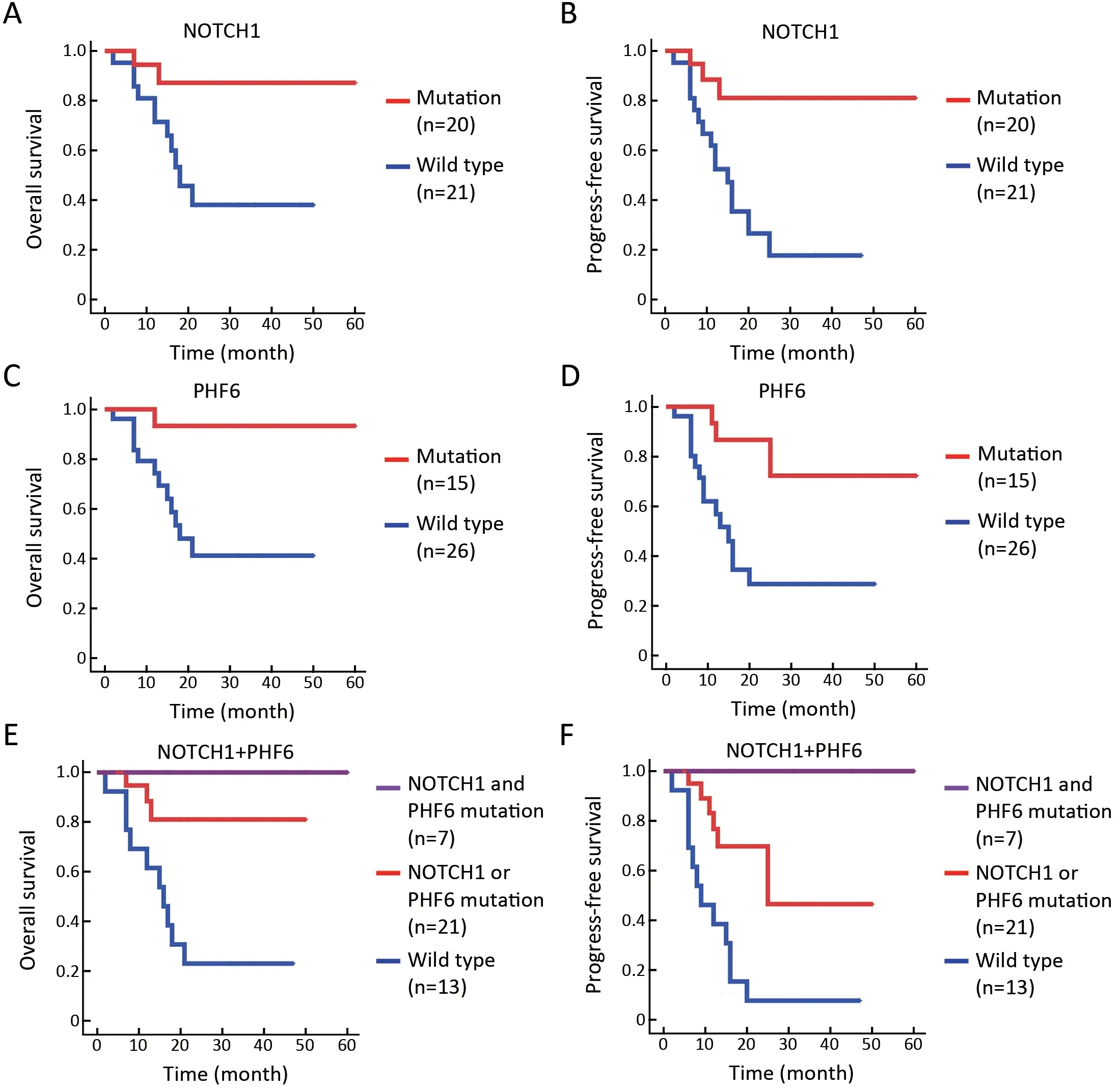
Figure 5 NOTCH1/PHF6 mutations predict favorable prognosis for patients with T-LBL.(A) OS (P=0.016) and (B) PFS (P=0.002) of TLBL patients with NOTCH1 mutations;(C) OS (P=0.009) and (D) PFS (P=0.004) of T-LBL patients with PHF6 mutations;(E) OS(P=0.001) and (F) PFS (P<0.001) of T-LBL patients with NOTCH1 and PHF6 mutations.Patients were divided into three groups:those with wild type for both NOTCH1 and PHF6 (n=13),those with mutations in either NOTCH1 or PHF6 (n=21),and those with mutations in both NOTCH1 and PHF6 (n=7).Survival curves are estimated with the Kaplan-Meier method and compared using a two-sided log-rank test.TLBL,T-cell lymphoblastic lymphoma;OS,overall survival;PFS,progression-free survival.
Discussion
To the best of our knowledge,this is the largest genomic analyses to date that revealed a comprehensive landscape of genetic alterations in T-LBL by whole-exome sequencing approach.In present study,32 genes were identified as driver genes.Although most of these genes have been previously reported in T-ALL,many of them have not yet been described in T-LBL (for example,JAK3,JAK1,RUNX1,WT1,etc.).Notably,some gene mutations were exclusively identified in T-LBL but not in T-ALL (for example,CDC27,MTRNR2L2,COL6A5andTMEM200C).Thus,we put forward that T-LBL might have a different genetic alterations profile that distinguishes it from TALL,although there was mutation overlap between the two.
The NOTCH signaling pathway plays a critical role in cell lineage commitment decisions during development(15).However,the central role of NOTCH1 in T cell transformation was only realized upon the identification of activating mutations in theNOTCH1gene present in over half of T-ALL cases (16).NOTCH1activating mutations were found in 48.78% of T-LBL in the present study(Supplementary Table S6).NOTCH1mutations typically involve specific domains responsible for controlling the initiation and termination of NOTCH signaling (17).In addition,FBXW7mutations,present in 14.6% of T-LBL cases,contribute to NOTCH activation by impairing the proteasomal degradation of activated NOTCH1 in the nucleus (18).
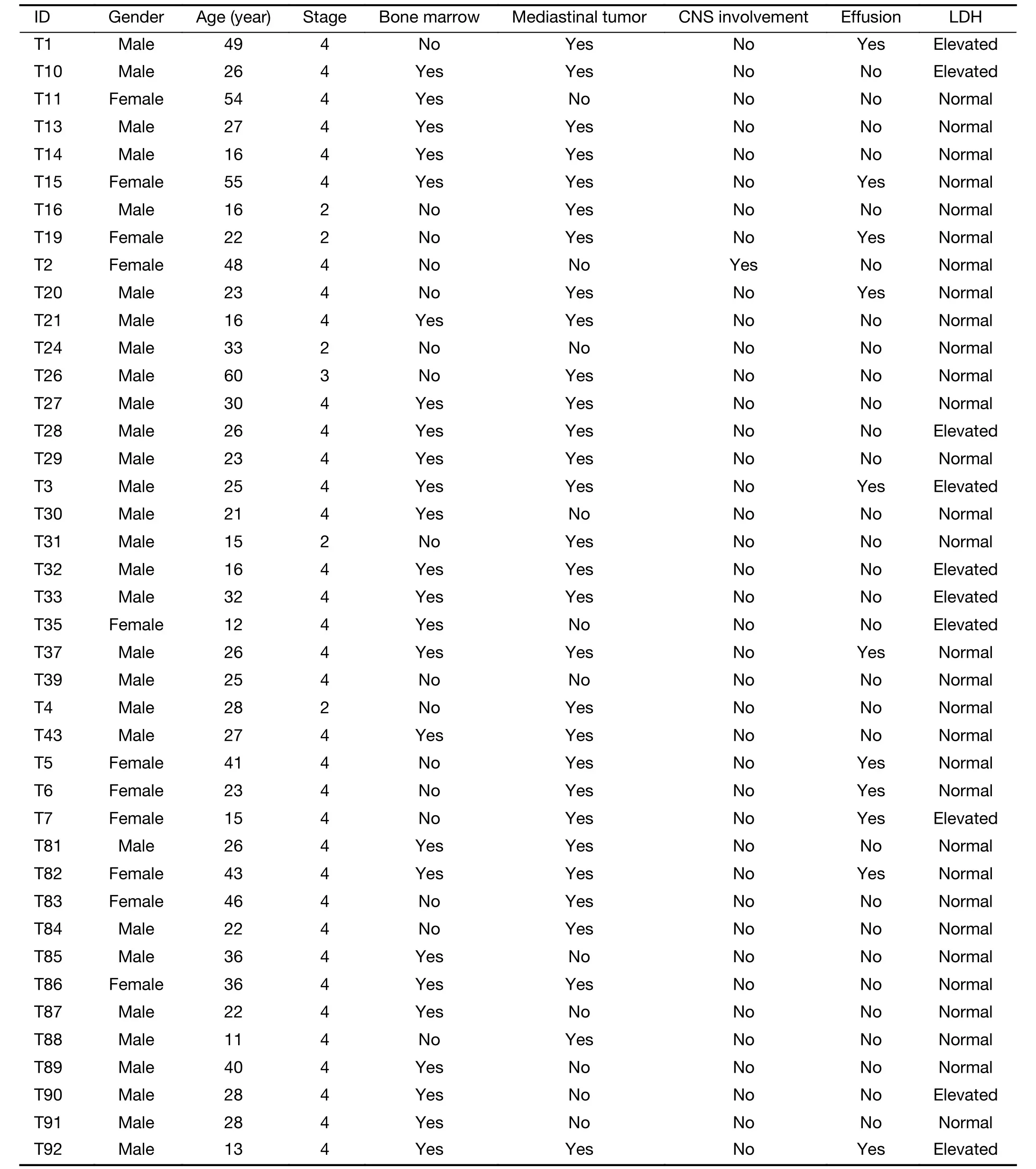
Table S1 Demographics and clinical features of 41 subjects with T-LBL
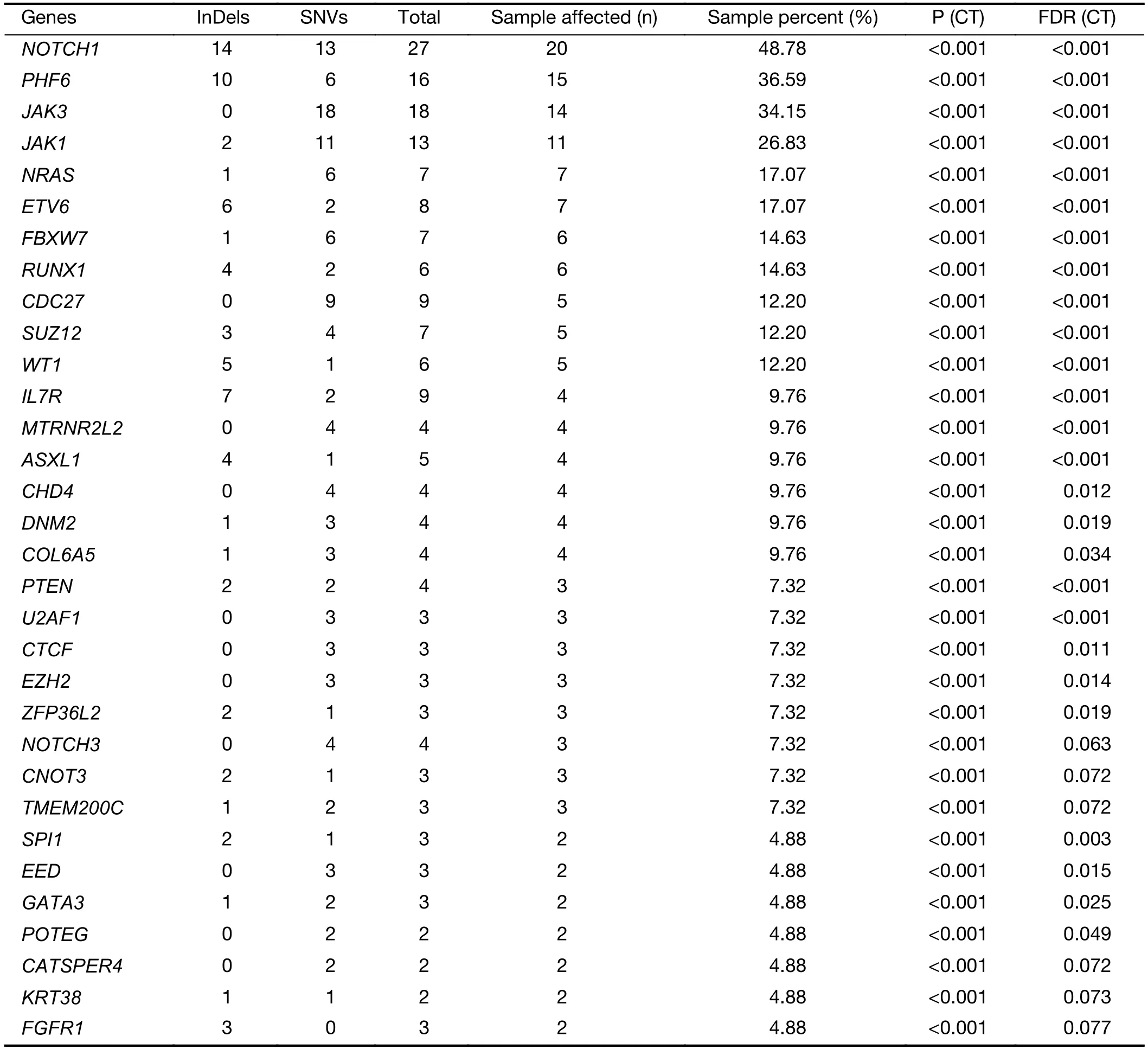
Table S2 Significantly mutated genes
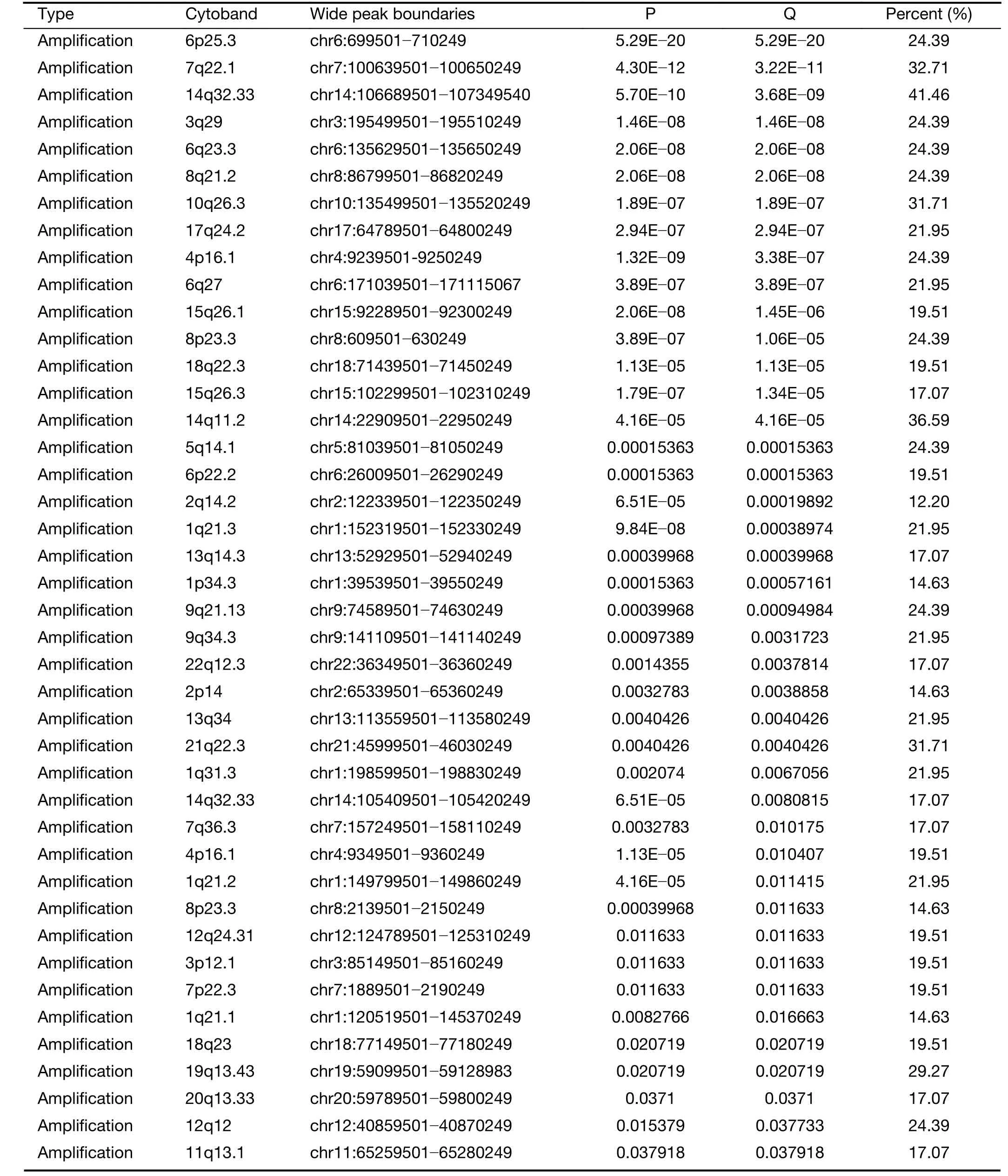
Table S3 Somatic copy-number alterations
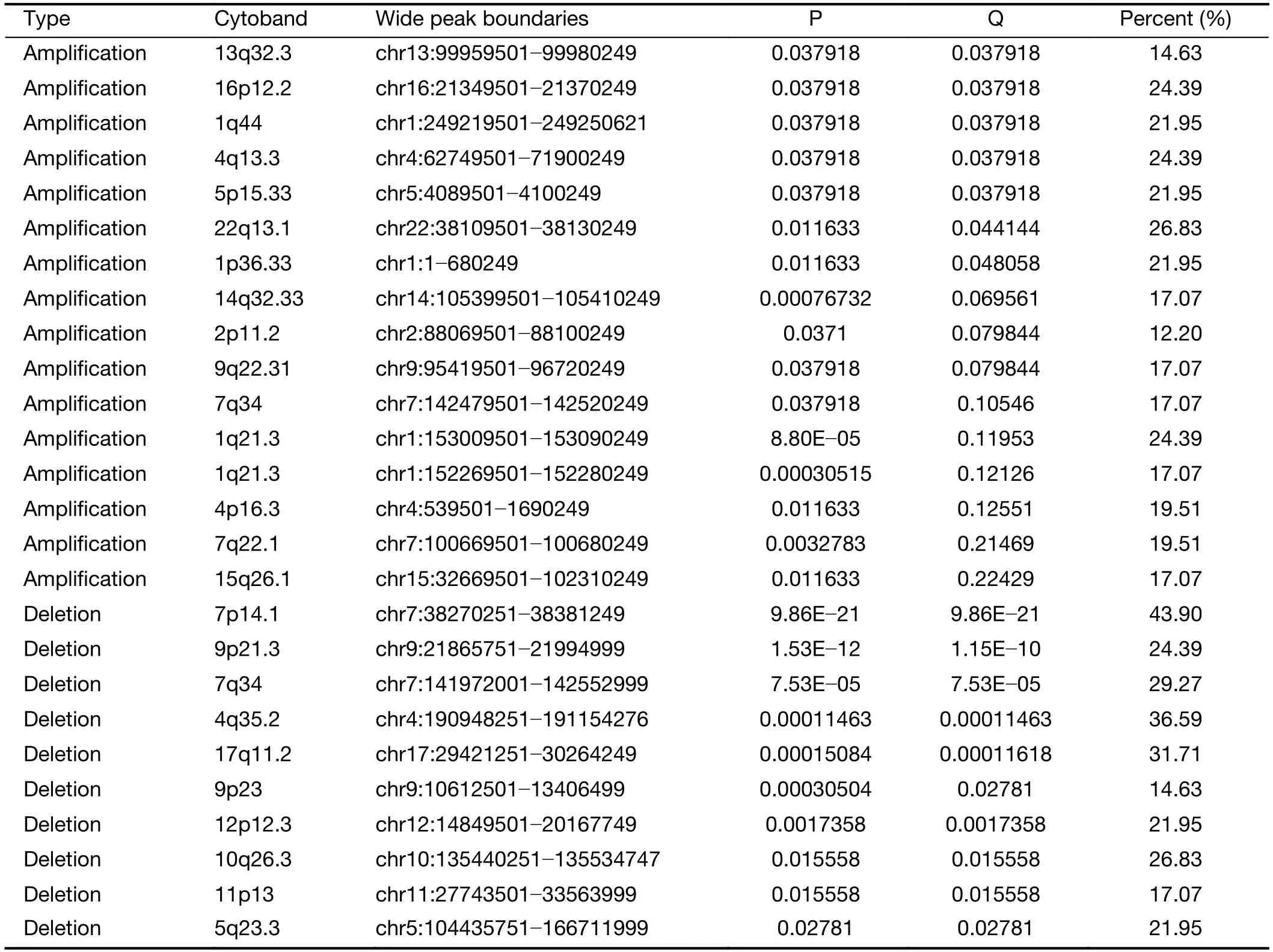
Table S3(continued)
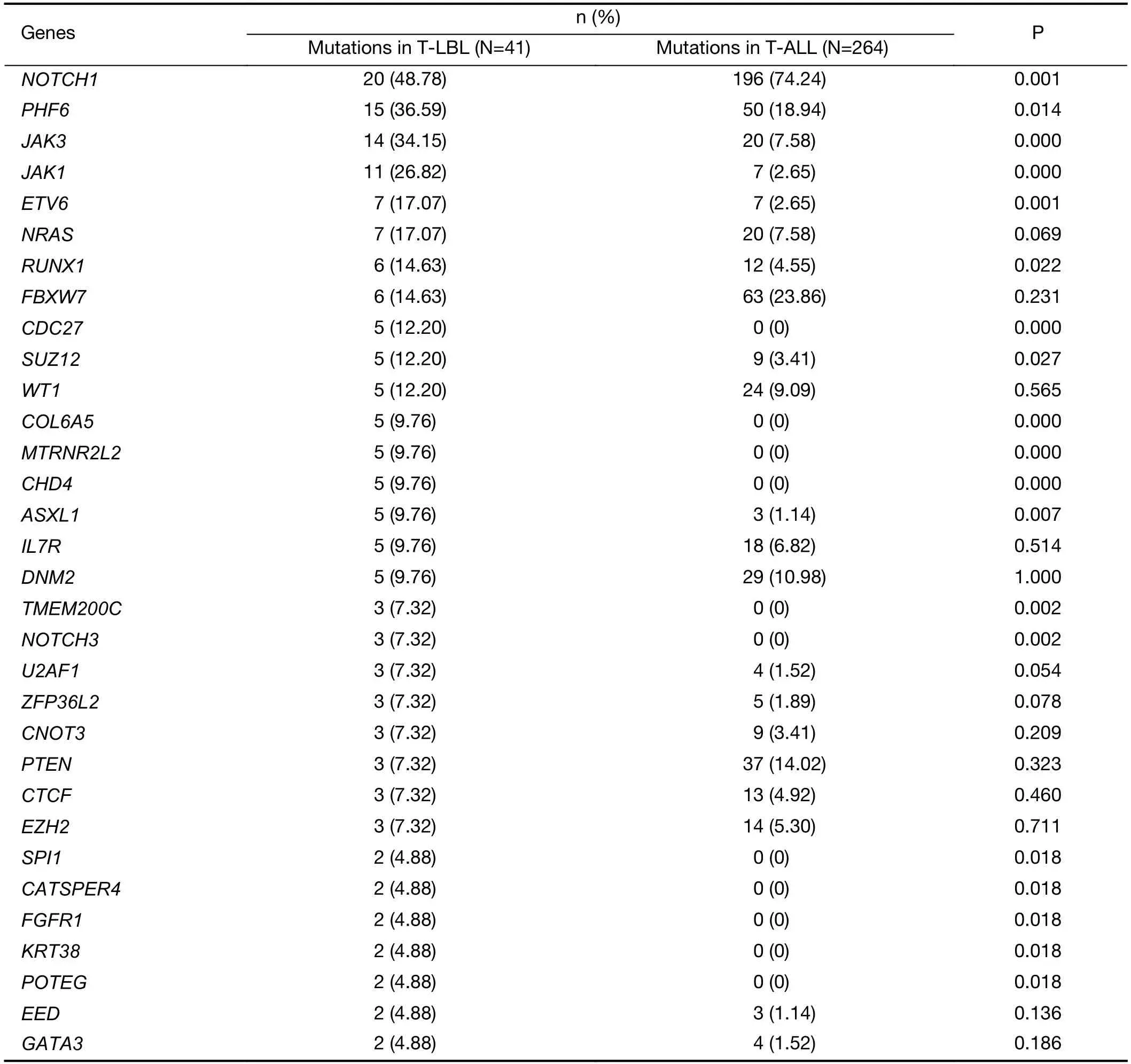
Table S4 Frequencies of commonly mutated genes between T-LBL and T-ALL
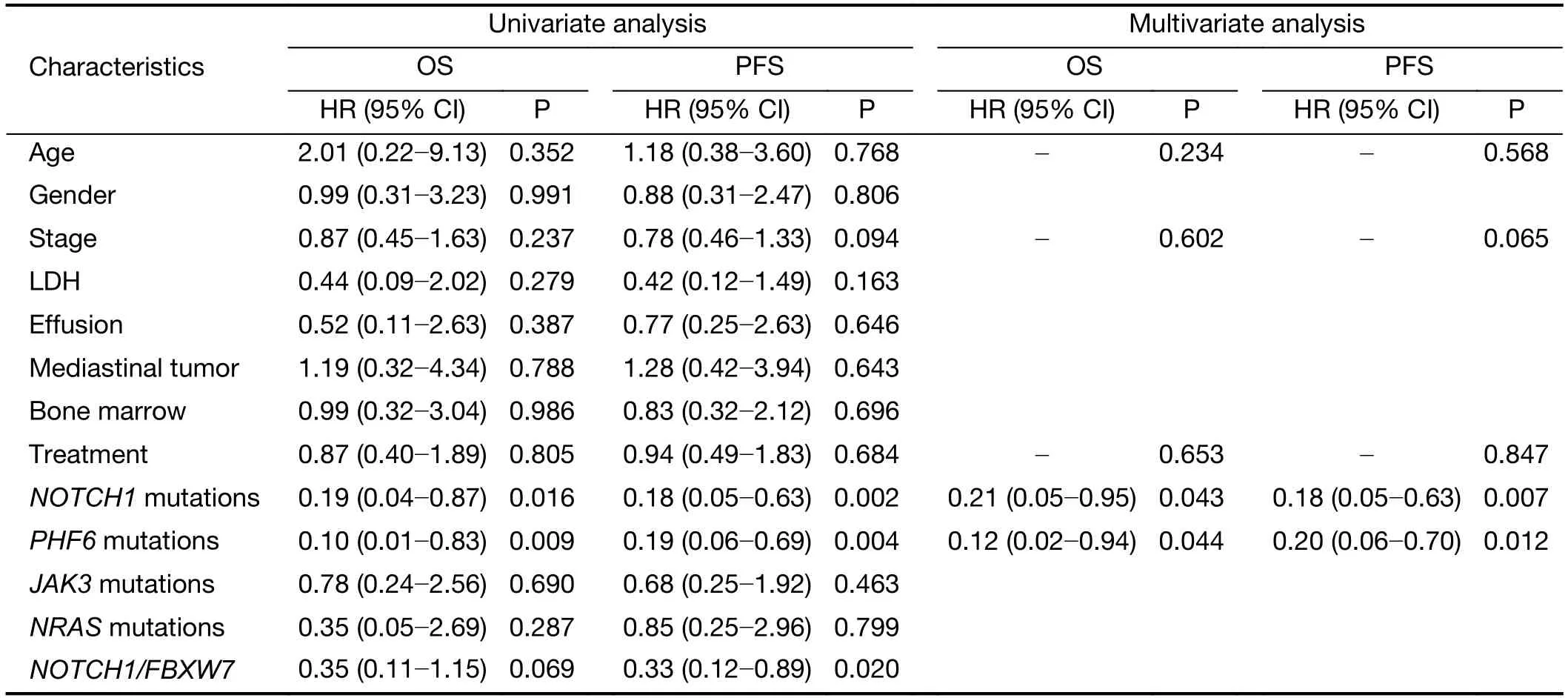
Table S5 Univariate and multivariate analysis for clinical and biological characteristics of survival in T-LBL (N=41)
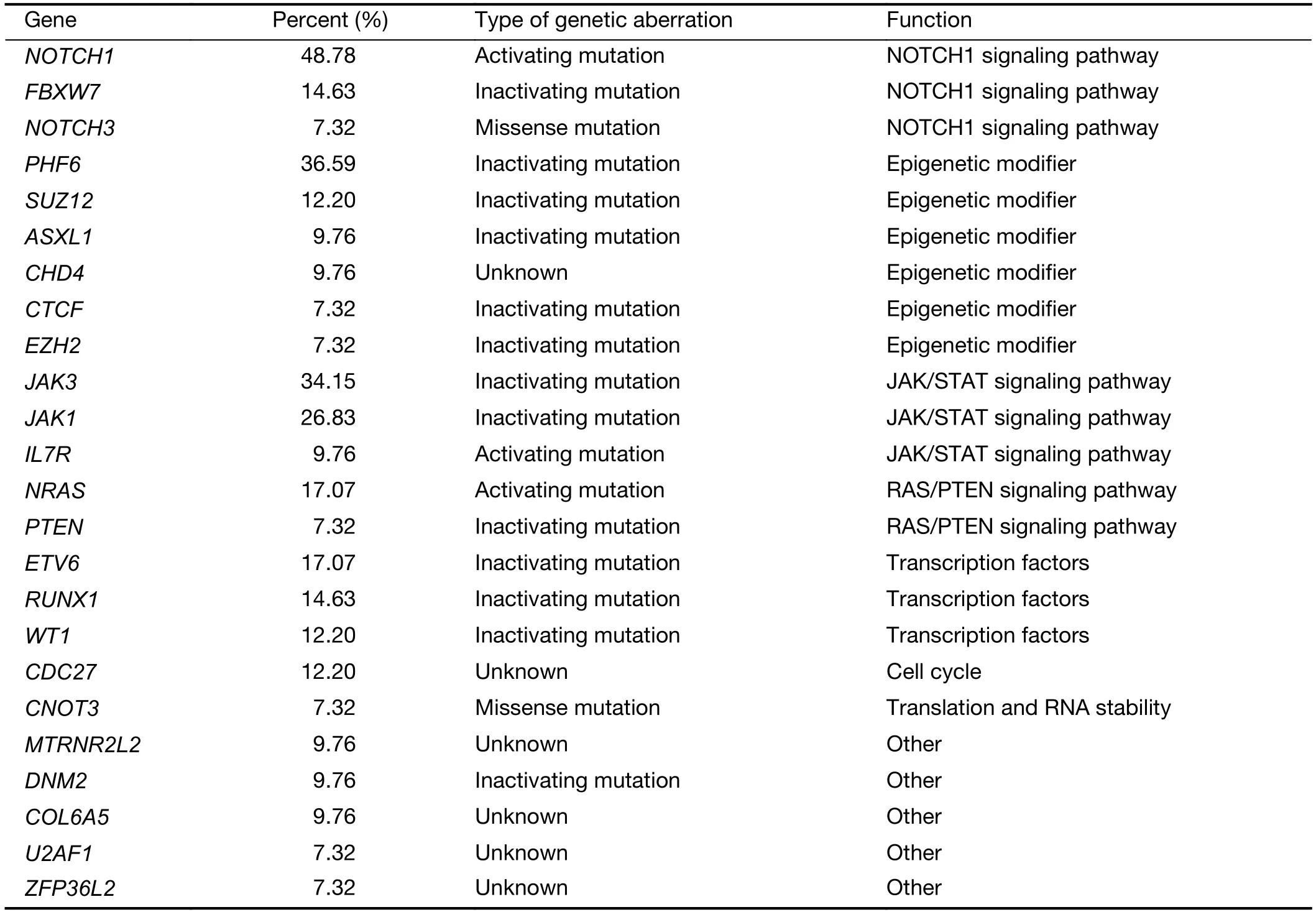
Table S6 Biological function of top gene mutations
As reported in T-ALL,mutations in epigenetic factors are also highly common in T-LBL.PHF6 is a highly conserved epigenetic transcriptional regulator,which is important for hematopoiesis and neurodevelopment (19).However,animal models have revealed that while PHF6 mutations/deletions may be initial events,they are insufficient for tumor initiation without additional driver mutations (20).Dysfunction of PHF6 usually cooperates with several other driver mutations in the development of leukemia.For example,inactivation of PHF6 in hematopoietic progenitors has been reported to facilitate NOTCH1-induced T-ALL,potentially through increasing leukemia-initiating cells and development of a “l(fā)eukemia stem cell transcriptional program” in lymphoblasts (21).Another study showed thatPHF6mutations frequently cooperate withHOX11L2overexpression orWT1mutations in pediatric T-ALL (22).Our study demonstrated that PHF6 mutations were particularly prevalent in T-LBL patients harboring mutations ofNOTCH1,which is consistent with previous reports in T-ALL (21).It suggests that PHF6 mutations may cooperate withNOTCHmutations during the oncogenesis of T-LBL.In addition,loss-of-function defects in the polycomb-repressive complex 2 (PRC2) components EZH2,SUZ12,and EED were also commonly found in present study.
The IL-7 receptor signals transmitted by JAK/STAT pathway are critical for the growth and survival of early T cell progenitor cells (23).Activating mutations inJAK1andJAK3have been reported in T-ALLs (24).Consistently,activatingJAK1andJAK3mutations were found in 26.8%and 34% of T-LBL cases,respectively.Moreover,somatic gain-of-function mutations in theIL7Rgene,encoding the IL7 receptor and resulting in constitutive activation of JAK/STAT signaling,were identified in 9.7% of T-LBL in our study.In addition to the JAK/STAT pathway,the RAS-MAPK and phosphatidylinositol 3-kinase (PI3K)pathways are also activated by IL7 that acts on the development of T cells.ActivatingNRASmutations and inactivatingPTENmutations occur in 17.0% and 7.3 of TLBL cases,respectively.
The transcription factor tumor suppressor genes have been shown to be mutated and deleted in T-LBL,includingETV6,RUNX1andWT1.ETV6encodes an ETS family transcriptional repressor strictly required for the development of hematopoietic stem cells (15).ETV6mutations encoding truncated proteins with dominantnegative activity were frequently found in early immature T-ALLs (15).Loss-of-function mutations inRUNX1occurred in 14.6% cases of T-LBL,suggesting a tumor suppressor role forRUNX1in T-cell transformation.As reported in T-ALL,WT1mutations found in T-LBL are predominantly heterozygous frameshift mutations resulting in truncation of the C-terminal zinc finger domains of this transcription factor (25).They are frequently associated with oncogenic expression of theTLX1,TLX3orHOXAoncogenes (25).
Although T-LBL shows many characteristics in common with T-ALL,there is still ongoing discussion on whether T-ALL and T-LBL are two distinct entities.To identify the critical molecular and cytogenetic differences that distinguish T-LBL from T-ALL,genetic alterations of TLBL and T-ALL were compared in our study.By integrating gene mutation and copy-number alteration data,we found that mutations in JAK-STAT and RAS signaling pathways were predominantly observed in T-LBL(58% and 34%,respectively),whereas NOTCH and cell cycle signaling pathways mutations were more prevalent in T-ALL.Therefore we believe that T-LBL might have a different genetic alteration profile that distinguishes it from T-ALL.This is also a finding of potential therapeutic relevance,providing a rationale for targeting these pathways in T-LBL cases.
T-LBL is commonly treated on T-ALL-derived protocols (4).Therapeutic stratification based on prednisone response and minimal residual disease assessment is well established in T-ALL but is not easy to extrapolate to T-LBL (26).Molecular genetic markers are promising candidates for risk stratification because they represent underlying biological properties of the subgroups,and their analyses can be standardized,which is essential for cooperative clinical trials.Hence the prognostic value of these genetic mutations in T-LBL was evaluated.Recent retrospective studies in T-LBL have reported association of outcome with loss of heterozygosity at chromosome 6q,biallelic T-cell receptor-γ deletions and with FBXW7 andNOTCH1mutations (27).Here we present the prognostic relevance of mutations in the genesPHF6andNOTCH1.In our study,we found that patients withNOTCH1mutations were associated with a favorable prognosis,which is consistent with previous researches.It has also been reported that co-existence ofNOTCH1andFBXW7mutations allowed identification of a low-risk subgroup of T-LBL with a more favorable prognosis thanNOTCH1alone (6);however,our study did not reproduce these findings.The discrepancy may be attributed to the population differences and sample size.We also found thatPHF6,an X-linked tumor suppressor gene (28),is the second most frequent mutated gene in T-LBL.Furthermore,the association ofPHF6mutations and survival in T-LBL was established for the first time.Mutation status ofPHF6was identified as another independent prognostic factor for improved survival of TLBL patients.Notably,the combination of mutation data ofPHF6andNOTCH1could allow identification of those patients who do extremely well on current T-ALL-derived protocols,but also a significant fraction of patients for whom treatment needs to be improved.Therefore,it might provide an alternative for early therapeutic stratification in T-LBL.
A limitation of this study is that we did not carry out whole-genome sequencing or RNA-sequencing for these cases,due to the difficulty of obtaining fresh T-LBL samples.Therefore,we can not identify gene rearrangements information in these cases,which might also have a critical role in the development of T-LBL,as it does in T-ALL (12).
Conclusions
We have identified multiple genes mutations and signaling pathways in T-LBL which are different from those in TALL,implying that T-LBL might have a different profile of genetic alterations distinguishing it from T-ALL.Consequently,this detailed portrait of the T-LBL genomic landscape offers a rationale for targeting these pathways in T-LBL.What’s more,the combination ofPHF6andNOTCH1mutations can be used to establish future molecular and prognostic analyses and early therapeutic stratification in T-LBL. Nevertheless,larger-scale independent prospective therapeutic trials are required to validate our findings.These studies not only advance current insights into the molecular basis of T-LBL,but may also have tangible clinical implications.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (No.U1904139 and 82070209).
Footnote
Conflicts of Interest:The authors have no conflicts of interest to declare.
 Chinese Journal of Cancer Research2022年2期
Chinese Journal of Cancer Research2022年2期
- Chinese Journal of Cancer Research的其它文章
- Pan-cancer tumor-infiltrating T cells:A great hallmark in cancer immunology research
- Current status and perspectives of conversion therapy for advanced gastric cancer
- CSCO guidelines for colorectal cancer version 2022:Updates and discussions
- Current situation of programmed cell death protein 1/programmed cell death ligand 1 inhibitors in advanced triplenegative breast cancer
- Establishment of an optimized CTC detection model consisting of EpCAM,MUC1 and WT1 in epithelial ovarian cancer and its correlation with clinical characteristics
- Prognostic and incremental value of computed tomographybased radiomics from tumor and nodal regions in esophageal squamous cell carcinoma