
ATP13A2 protects dopaminergic neurons in Parkinson's disease: from biology to pathology
2022-04-14 06:33:38TaoDangWenJingCaoRongZhaoMingLuGangHuChenQiao
Tao Dang, Wen-Jing Cao, Rong Zhao, Ming Lu, Gang Hu, Chen Qiao,?
1Department of Clinical Pharmacy, the Affiliated Hospital of Jiangsu University, Zhenjiang, Jiangsu 212001, China;
2College of Pharmacy, Jiangsu University, Zhenjiang, Jiangsu 212013, China;
3Jiangsu Key Laboratory of Neurodegeneration, Department of Pharmacology, Nanjing Medical University, Nanjing,Jiangsu 211166, China;
4Department of Clinical Pharmacy, Xiangtan Central Hospital, Xiangtan, Hunan 411100, China.
Abstract As a late endosomal/lysosomal transport protein of the P5-type, ATP13A2 is capable of removing the abnormal accumulation of α-synuclein, which maintains the homeostasis of metal ions and polyamines in the central nervous system. Furthermore, ATP13A2 regulates the normal function of several organelles such as lysosomes,endoplasmic reticulum (ER) and mitochondria, and maintains the normal physiological activity of neural cells.Especially, ATP13A2 protects dopaminergic (DA) neurons against environmental or genetically induced Parkinson's disease (PD). As we all know, PD is a neurodegenerative disease characterized by the loss of DA neurons in the substantia nigra pars compacta. An increasing number of studies have reported that the loss-offunction of ATP13A2 affects normal physiological processes of various organelles, leading to abnormalities and the death of DA neurons. Previous studies in our laboratory have also shown that ATP13A2 deletion intensifies the neuroinflammatory response induced by astrocytes, thus inducing DA neuronal injury. In addition to elucidating the normal structure and function of ATP13A2, this review summarized the pathological mechanisms of ATP13A2 mutations leading to PD in existing literature studies, deepening the understanding of ATP13A2 in the pathological process of PD and other related neurodegenerative diseases. This review provides inspiration for investigators to explore the essential regulatory role of ATP13A2 in PD in the future.
Keywords: ATP13A2, Parkinson's disease, dopaminergic neurons, lysosome, α-synuclein
Introduction
Parkinson's disease (PD) is a complex neurodegenerative disease characterized by the loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNc), coupled with lysosomal dysfunction and the accumulation of Lewy body in the central nervous system (CNS). Symptoms include motor symptoms such as muscle rigidity,bradykinesia, restlessness, tremor, and abnormal gait,and non-motor symptoms such as depression, anxiety,hypotonia, sleep disorders, affective disorders andhallucinations[1].ATP13A2(also calledPARK9)gene encodes a transmembrane lysosomal P5-type ATPase (ATP13A2). Previous studies have shown that the loss-of-function of ATP13A2 plays a significant role in the pathologic progression of PD.
ATP13A2 protein is comprised of five different domains with corresponding specific functions[2].Mutations in different domains lead to different dysfunction outcomes, such as the abnormal aggregation of α-synuclein, metal ion injuries,polyamine homeostasis damage, glycolysis processes disorder and destroyed autophagy processes, and then culminate in lysosome, mitochondria, and endoplasmic reticulum (ER) dysfunction[1,3]. Moreover, the disruption of cellular homeostasis caused by ATP13A2 mutation will further stimulate microglia and astrocytes to release inflammatory factors, which increases neuroinflammation. It has been reported that the regulation of ATP13A2 in the processes of αsynuclein, metal ion homeostasis and autophagy may serve as a promise target for the development of drugs for PD[4]. Other studies have suggested that ursolic acid and chlorogenic acid can protect neural cells against inflammatory risk and mitochondrial oxidative stress, but the mechanism of their interaction with ATP13A2 remains unclear[5].
Therefore, with an in-depth understanding of the physiological structure and function as review, we described the possible molecular mechanism of PD based on the structure and function of ATP13A2, in order to find the root cause of pathological changes induced by ATP13A2 mutation.
Normal structure of ATP13A2
ATP13A2 is a lysosomal transporter that comprises 1180 amino acids and 29 exons[6]. The protein has 10 transmembrane helical domains, from M1 to M10. Of which, M4, M5, M6, and M8 are located at the core transmembrane region, while M1, M3, M7, M9, and M10 are at the outer domains[7–10]. In the hydrophobic section of the N-terminal domain, an additional transmembrane helical structure is divided. The Nterminal domain does not cross the membrane but remains on the surface of the cytoplasmic membrane[7,10–12](Fig. 1).
Recent studies demonstrated that the M4 fragment,as the core domain, is located at the middle of the transmembrane segment, and contains a hydrophobic PP(A/V)LPAx sequence motif, while M5 has an amine side chain group[10,13]. In addition, ATP13A2 has three highly specific subdomains, including a nucleotide-binding (N) domain, a phosphorylated (P)domain, and a conserved cytosolic actuator (A)domain[14]. The P domain contains aspartic acid residues which are involved in the formation of phosphorylase intermediates, and the braking mechanism domain (A) is mainly responsible for the dephosphorylation of the P domain, while the N domain is proximal to the P domain in the phosphorylation state[7,10]. In addition, residues in the core transmembrane region are involved in the formation of ligand binding sites[10](Fig. 1). Different residues enable ATP13A2 to maintain the homeostasis of heavy metal ions, proteins and polyamines to protect the physiological functions of organelles such as mitochondria, ER and lysosomes.
Normal function of ATP13A2
As a member of the P5 ATPase transporter family,ATP13A2 is mainly localized to lysosomes[15].Researches have shown that ATP13A2 maintains neuronal healthy state by protecting the homeostasis of metal cations such as Fe3+, Mn2+, Zn2+, and Ca2+,increasing the elimination of excess α-synuclein and polyamines, and even maintaining the function of organelles such as lysosome, ER and mitochondria[12].
Prevention of metal ion-induced toxicity
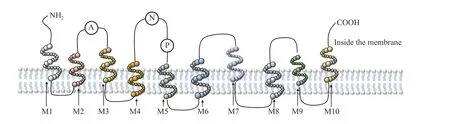
Fig. 1 The structure of ATP13A2. ATP13A2 contains 10 transmembrane domains M1 to M10, of which M4, M5, M6, and M8 are the core transmembrane regions. A, P and N respectively represent the three subdomains of ATP13A2. A is the conserved cytosolic actuator domain; P is the phosphorylation domain; N is the nucleotide binding domain.
Metal ions play vital roles in the physiological process of cells. For example, as a cofactor of various enzymes, Mn2+is an indispensable metal cation to maintain the normal physiological process of various organelles[15]. Studies have shown that Zn2+plays a significant role in various biological pathways, such as cell signal transduction, enzyme catalysis, immune function and protein stabilization[16]. An appropriate amount of metal ions is contributed to the normal development of various physiological processes.However, if the number of metal ions in cells exceeds a certain amount or the homeostasis is destroyed, it may negatively influence the physiological processes of neuronal metabolism and even induce the death of DA neurons.
It has been reported that ATP13A2 is not only a metal cation transporter, but it regulates the homeostasis of various metal ions through its own non-transporter function, so that neurons could avoid toxicity[17–18]. ATP13A2 in non-lysosomal vesicles can recognize excessive Zn2+and increase its transport rate by promoting the entry of Zn2+into vesicles, so the excessive Zn2+exclusion can be achieved to maintain Zn2+homeostasis and prevent Zn2+toxicity[19].
Furthermore, lysosomes are the major repositories of chelate iron and calcium (Ca2+)[20]. The lysosomal exocytosis process requires the rapid increase of Ca2+on its surface, and ATP13A2 regulates lysosomal exocytosis by regulating Ca2+homeostasis. At the same time, in lysosomes, Ca2+homeostasis is maintained by controlling exocytosis, and excessive Ca2+can be excluded from the cells by exocytosis,thus avoiding the excessive Ca2+-induced mitochondrial oxidative stress[21–22].
Fe3+promotes the accumulation of α-synuclein and induces mitochondrial dysfunction, thereby inducing cell death. For Fe3+homeostasis, ATP13A2 regulates iron homeostasis by affecting sorting connexin 3 reverse transcriptase-mediated iron transporter recirculation. On the other hand, ATP13A2 protects lysosomal exocytosis and intracellular transport processes by preventing Fe3+-induced remodeling of the cytoskeleton[20,23–24], and even protects DA neurons from iron-induced cytotoxicity by maintaining the integrity of lysosomal membrane.
ATP13A2 protects neurons from Mn2+-induced toxicity by maintaining Mn2+homeostasis through transporting Mn2+into the lysosome, which is then excreted in the form of vesicle fusion[15,25–26]. In addition, ATP13A2 also promotes cell uptake of polyamines, which can chelate a variety of metal cations to prevent metal ion toxicity and provide protection for cells. (Fig. 2B)[27].
Prevention of the aggregation of α-synuclein
As a naturally unfolded and easily aggregated protein, α-synuclein also plays an important role on neural cells, such as affecting the storage of Mn2+[26,28].Lewy bodies are hallmark material of PD and are composed of aggregated α-synuclein molecules. This agglomeration of α-synuclein causes protein toxicity,organelle dysfunction, and even DA neuronal dysfunction[29–30]. Previous studies demonstrated that ATP13A2 promotes the degradation of α-synuclein through autophagy, and secrets α-synuclein to the extracellular level, preventing the toxicity caused by α-synuclein aggregation[31]. Firstly, ATP13A2 regulates the ubiquitin-proteasome system (UPS) and autophagy-lysosomal mechanisms for the recognition and degradation of abnormally aggregated α-synuclein[26,32–33]. Secondly, ATP13A2 regulates the polymerization of α-synuclein on the inner membrane and enhances its binding to the membrane, while simultaneously controls the number of vesicles. The aggregation of α-synuclein in intima increases the amount of α-synuclein in vesicles, and the increasing number of vesicles can carry more α-synuclein.Regulation of exosome biogenesis by ATP13A2 promotes α-synuclein secretion from vesicles into the extracellular space[34]. In addition, secreted extracellular α-synuclein can be absorbed and degraded by astrocytes, thus protecting neurons from the toxicity of extracellular α-synuclein (Fig. 2E–F)[35].
Regulation of intracellular polyamine homeostasis
Polyamines are abundant aliphatic polycations,which play a prominent role in cell proliferation,differentiation, apoptosis, post-translational modification of proteins and regulation of ion channels[27].However, high concentration of polyamines produces cytotoxicity, which is not conducive to the normal physiological process of neurons, or even lead to DA neuronal degeneration[36]. Studies have shown that ATP13A2 is a transporter of lysosomal polyamines,which can transport spermine and spermidine from lysosomal to cytoplasm to supplement intracellular polyamines[27]. Surprisingly, ATP13A2 can promote the uptake of polyamines through endocytosis.Endocytic polyamines are stored in lysosomes and supplemented into cells when necessary, through the transport function of ATP13A2. In addition, ATP13A2 can also prevent lysosomal dysfunction and lysosomal rupture caused by accumulated polyamines[37–38].ATP13A2 maintains the homeostasis of total cellular polyamines and prevents cytotoxicity resulting from overproduction (Fig. 2B).
Regulation of autophagy

Fig. 2 The function of ATP13A2 mutations in neural cells. A: ATP13A2 regulates cellular uptake of polyamines and controls the intracellular distribution of polyamines in lysosomes. B: ATP13A2 mutations cause lysosomal rupture and intracellular accumulation of polyamines and metal ions, causing mitochondrial oxidative stress. C and D: Autophagosomes proceed under the regulation of ATP13A2,and fused with lysosomes to form autolysosomes to complete autophagy, then mutations lead to impaired autophagic process. E and F:Aggregation of α synuclein expelled from neurons are taken up and degraded by astrocytes, while the ATP13A2 mutation affects the process causing inflammatory responses in astrocytes and damage to neurons. G: ATP13A2 mutations resulted in abnormal α-synuclein aggregation and ER membranes, meanwhile the mutant ATP13A2 binds the aggregated α-synuclein and co-locate on the ER. SYT11: synaptotagmin-11;SPM: spermine; α-syn: α-synuclein; ER: endoplasmic reticulum.
As mentioned above, ATP13A2 inhibits the aggregation of α-synuclein and prevents protein toxicity by regulating the autophagy process. In addition, ATP13A2 eliminates damaged organelles by regulating autophagy process and maintaining normal operation of mitochondria and lysosomes. Firstly,ATP13A2 regulates Synaptotagmin-11 (SYT11)expression by modulating the activity of SYT11 mRNA, thus regulating the autophagy process(Fig. 2D)[39]. Furthermore, ATP13A2 also facilitates autophagy by promoting lysosomal-autophagosome fusion. Previous studies reported that ATP13A2 promoted the degradation of insoluble proteins by increasing the activity of α-tubulin deacetylase(HDAC6). ATP13A2 also locates HDAC6 to lysosomes and promotes the fusion of lysosomes and autophagosomes to complete autophagy (Fig. 2C)[40].In summary, ATP13A2 removes abnormal molecules and damaged organelle, ensuring the intracellular autophagic homeostasis and maintaining the normal function of DA neurons.
ATP13A2 mutation and PD
Emerging research has shown that neuronal degeneration in PD is associated with mistranslation caused by mutations in LRRK2, PINK1, PLA2G6,GBA and ATP13A2[41]. Mutations in ATP13A2 lead to Kufor-Rakeb syndrome, which is an atypical autosomal recessive form of PD in adolescents[42]. The functional loss of mutated ATP13A2 affects various areas of the CNS, such as the cortical pyramidal system, extrapyramidal system, brainstem cerebellum,and the peripheral nervous system, leading to parkinsonism.
ATP13A2 mutations at different loci induce various Parkinson's symptoms. In Shen's study,ATP13A2gene in patients with PD was analyzed and found to be mutated in different degrees. The frequency of T allele in c.1815C>T, c.2637C>T, c.3192C>T and A allele in c.2970G>A, c.3516G>A was significantly higher in PD patients compared to the healthy group[1].In Fonzo's research, a 12-year-old Brazilian adolescent with PD presented with levodopa-induced motion fluctuations, dyspraxia, severe hallucinations,and supranuclear vertical gaze paralysis. A mistranslated mutation (Gly504Arg) was identified in the ATP13A2 sequence[43]. In the ATP13A2 sequence of two disaffected siblings from Turkey, a framecoding deletion in exon 15 (c.1422_1423del:p.474fs)putatively lead to a premature termination codon,resulting in the loss of nitrogen and phosphorus domains which leads to PD[44]. In addition, an 18-yearold male patient from Pakistan developed hyperreflexia and spastic gait, whoseATP13A2gene was found mutated (c.2218C>T; p.Arg740Ter)[42].Mutation in exon 22 ofATP13A2gene was found in a 16-year-old clinical patient who presented with hand tremor, gait disorder, and psychiatric symptoms of restlessness and nervousness[45]. An 18-year-old boy with developmental delays learned to walk at age 3,started talking at age 5, and developed PD symptoms of gait disturbance, unsteady walking, and cognitive difficulties at age 11. Clinical investigations revealed reduced dopamine transport in his brain. The p.Arg740Ter mutation in theATP13A2gene was identified in the genetic analysis of this boy. The above cases showed that ATP13A2 mutation can induce PD and the mutation sites were diverse.
In addition, ATP mutations are highly correlated with genetics and development. In one case, the patient's parents and his siblings were healthy and did not show symptoms associated with PD. His father had the recessive mutation c.1321A>T inATP13A2and his mother had the recessive mutation c.3205G>A inATP13A2. Patients with PD present in infancy with developmental delays, problems with limb coordination, inability to care for themselves, and learning difficulties, dysarthria, limb stiffness and tremors[46]. However, in another family with PD, the patient's parents carried the mutated ATP13A2 heterozygote p.Q648X, but both of them were healthy.The patient, a 27-year-old male, presented at age 16 with symptoms of PD including abnormal gait,dysarthria, and dysphagia. His brother had similar symptoms. Genetic analysis revealed that the brothers had the pathogenic p.Q648X mutation of ATP13A2[47].
The analysis of clinical symptoms and genes of patients with PD showed thatATP13A2mutation plays a significant role in PD, and the gene mutations are diverse, and the degree of influence and onset time of patients remain uncertain. It is of great significance to explore the genetic and environmental risks of ATP13A2 mutation leading to PD. Presently, the exact pathological mechanism of PD induced byATP13A2gene mutation has not been fully elucidated.Previous research suggests that mutation of the key gene locus of ATP13A2 leads to impaired metal cation homeostasis in neural cells, and induces the protein retention and degradation in ER, leading to the degeneration and even the death of DA neurons,which may be the main reasons for PD pathological changes[48].
Characterization of animal models with ATP13A2 mutation
Several studies described animal models induced by ATP13A2 mutation to simulate human PD-like symptoms so as to deeply analyze the pathological mechanism of ATP13A2 mutation-induced PD, and try to bring novel targets for PD drug therapy.
Mice with the lack of ATP13A2 showed senescence, mitochondrial dysfunction, lysosomal functional block and increased ultrastructural features,as well as aggregation of α-synuclein in neurons[49].The mouse model showed motor deficiency, glial hyperplasia, and lipofuscin deposition. Furthermore,mice with ATP13A2 mutation had age-dependent autophagy damages. When they got older, weight loss,liver enlargement, and adipose tissue volume decrease were observed[40]. In addition, after intraperitoneal injection of manganese chloride for ATP13A2 knockout mice, high levels of accumulation of Mn2+and Fe3+were found in the brain, so was the aggregation of α-synuclein in SNc[15]. There was a report that ATP13A2 mutants lacking ATPase activity in the brain resulted in the degeneration and motor dysfunction of DA neurons[50].
HumanATP13A2gene and theCaenorhabditis elegans(C.elegans)CATP6gene show similar homology[20]. Researchers have usedC. elegansas an animal model with ATP13A2 mutation. The loss of Catp6 in theC. elegansshowed misfolded and aggregated α-synuclein in DA neurons. The models exhibited serious motility defects, such as significantly reduced shaking rate and egg hatching time[51–52]. Moreover, theC. elegansharboring ATP13A2 mutation was more sensitive to Fe3+and rotenone-induced oxidative stress and respiratory deficiency[20]. Additionally,C. elegans' growth ability was inhibited in the polyamine environment, showing as the shortened length of the stunted worms, and the more serious situation inC. elegansmodels in lack of ATP13A2 homologs[37].
These models suggest that ATP13A2 plays an important role in the development of PD. Exploring the exact pathological mechanism of PD induced by ATP13A2 mutation is expected to provide new ideas for elucidating the pathological mechanism of PD and accelerate the process of drug development.
The potential mechanism of ATP13A2 in PD pathology
Previous reports suggest that ATP13A2 may provide protection against genetic and environmental factors that contributed to PD[37]. However, ATP13A2 protein needs to be activated to exert a neuroprotective effects against PD[53]. Under normal circumstances,the N-terminal node domain of ATP13A2 appears to block ATP13A2 activity, thus requiring phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] and phosphatidic acid (PA) to bind to the N-terminal of ATP13A2 to stimulate ATP13A2 phosphorylation[33,53]. There are three specific lipid binding sites at the N-terminal end of ATP13A2, including LBS1,LBS2 and LBS3, in which PI(3,5)P2 binds to LBS2,and PA binds to LBS3, through which, the protective effects of ATP13A2 are awakened. The inhibition of N-terminal activity is reduced due to the specific binding of both lipids to the N-terminus, thereby activating the protective effects of ATP13A2[11,54].Furthermore, the protective effects of ATP13A2 are exerted by the stimulation of PI(3,5)P2 and PA in the autophosphorylation of ATP13A2 through binding to the N-terminal, thus assisting DA neurons to resist cytotoxicity induced by metal ions and mitochondrial damage. And ATP13A2 promotes the fusion of lysosomes and autophagosomes to form an autophagy-lysosome mechanism, removing damaged mitochondria, other organelles, and abnormal proteins[40]. Besides, protein ubiquitination is a prerequisite for protein recovery and degradation, and activated ATP13A2 appears to promote protein ubiquitination to degrade abnormally aggregated αsynuclein[32,34]. ATP13A2 also promotes glycolysis activity by maintaining metal ion homeostasis,ensuring the normal operation of glycolysis function,and alleviating the mitochondrial stress-induced damage through glycolysis mechanism[55]. In general,ATP13A2 provides protection against genetic and environmental risk factors for PD through glycolysis,phosphorylation, ubiquitin-protease and autophagylysosomal mechanisms.
Effects of ATP13A2 mutation on lysosomes
Lysosomes are reservoirs for a variety of proteolytic enzymes and heavy metal cations, and are the main routes for the intracellular distribution of polyamines[20,27,56]. ATP13A2 is a lysosomal transporter and mainly locates to lysosomes.Functional loss caused by ATP13A2 mutation affects normal function of lysosomes[15]. Previous studies indicated that ATP13A2 regulated the homeostasis of metal ions, and its mutation interfered with Mn2+,Zn2+, Ca2+, Fe3+, and other metal ion homeostasis.Since Mn2+is a cofactor for several enzymes,ATP13A2 mutation can affect the activities of various hydrolases in lysosomes and thus induce lysosomal dysfunction[21]. It was also reported that Zn2+dysregulation reduced the activity of lysosomal hydrolase. We believe the effects of metal ion homeostasis dysregulation on lysosomal hydrolase activity lead to α-synuclein aggregation caused by ATP13A2 mutation[41]. In addition, Ca2+and Fe3+play key roles in maintaining the integrity of lysosomal membrane, lysosomal exocytosis, and other transport processes. The homeostasis damage caused by ATP13A2 mutation will lead to the damage of lysosomal exocytosis and the integrity of lysosomal membrane, eventually culminating in lysosomal dysfunction[21,23]. Lysosomes are effective polyamines exporters, thus loss of ATP13A2 facilitates polyamine accumulation in lysosomes, lysosome breakdown, and the aggravation of α-synuclein polymerization and aggregation, leading to oxidative stress in mitochondria[38]. Besides, in a circuitous manner, αsynuclein polymerization also causes lysosome dysfunction[34]. Therefore, in addition to the direct influence of ATP13A2 mutation on lysosomes, other organelle abnormalities caused by ATP13A2 mutation may also affect the normal function of lysosomes.Homeostasis disruption induced by one factor will lead to changes in homeostasis and function of other substances, and the corresponding interactions between these changes may aggravate lysosomal dysfunction and lysosomal rupture.
Effects of ATP13A2 mutation on mitochondria
ATP13A2 mutation impairs the lysosomal function and homeostasis of metal ions and polyamines,inevitably impacting the mitochondrial function. The impaired polyamine homeostasis caused by ATP13A2 mutation will lead to the accumulation of reactive oxygen species (ROS) in mitochondria, causing oxidative stress[24,38]. In addition, the impaired homeostasis of Fe3+and Zn2+decreases the mitochondrial membrane potential and increases ROS,causing oxidative stress[20,50,57]. ATP13A2 mutants and aggregated α-synuclein act together on the mitochondria, inducing oxidative stress and abnormal mitochondrial functions[29]. These actions have reduced the production of energy and are detrimental to the normal function of cells. Besides, ATP13A2 mutation impairs the functions of lysosome and ER which will affect the synthesis of proteins and enzymes. Glycolytic enzyme changes due to ATP13A2 mutation, resulting in impaired glycolytic function, which will aggravate mitochondrial oxidative stress and lead to mitochondrial dysfunction[55,58]. Therefore, the effects of ATP13A2 mutation on mitochondria are complex and multifactorial, and more studies are needed to clarify them.
Effects of ATP13A2 mutation on ER
ER is an important organelle in protein processing and all proteins need to be processed by ER.ATP13A2, as a type of P5B ATPase, is also treated by ER. ATP13A2 mutations cause transcription abnormalities, thus when passing through the ER, the organelle cannot properly process the mutated protein,resulting in its inability to leave the ER. These affect the normal ER function, leading to the influence of other proteins such as lysosomal hydrolase, thus reducing lysosomal-mediated autophagosome clearance, and inducing polymerization and misfolding of α-synuclein. The mutant ATP13A2 binds to α-synuclein aggregates and increasing protease K resistance to α-synuclein aggregates. In addition, the mutant ATP13A2 and aggregated αsynuclein co-locate in the abnormal ER, forming abnormal ER structures, and impairing functional outputs[29].
Effects of ATP13A2 mutation on autophagy
As described, mutations in ATP13A2 affect the activity of hydrolases in lysosomes and autophagy process. The functional loss of mutated ATP13A2 induces ubiquitination and degradation of SYT11,leading to a decreased SYT11 level. Since ATP13A2-mediated autophagy is dependent on SYT11,decreased SYT11 levels can cause autophagy dysfunction, lysosomal insufficiency, and autophagy blockade[39]. Furthermore, ATP13A2 mutations also impair lysosomal-autophagosome fusion, resulting in impaired insoluble protein degradation and clearance,and even mitochondrial damage[40].
Effects of ATP13A2 mutation on neuroinflammation
Glial cells are widely present in CNS, and both microglia and astrocytes are widely distributed in the brain. As an innate immune cell, microglia produce inflammatory cytokines resisting malicious stimuli,then protecting DA neurons. If prolonged stimulation leads to excessive inflammation of microglia, it can induce degeneration of neurons and neurotoxicity.Astrocytes mainly regulate the homeostasis of neural cells and maintain the normal biological function of DA neurons. In addition, there are neurotrophic factors in astrocytes, which provide energy for the metabolism of neurons. Astrocytes also exhibit the capacity for inflammatory response and immune regulation. When stimulated, astrocytes instantly activate and release inflammatory factors to damage neurons[59].
As previously stated, ATP13A2 mutation may not directly cause neuroinflammation. ATP13A mutations lead to abnormal α-synuclein aggregation, oxidative stress in mitochondria, impaired metal ion homeostasis and metal ion toxicity. The aggregation of α-synuclein caused by ATP13A2 not only produces proteotoxicity, but also stimulates and activates microglia, causing autophagy defects in microglia, and releasing the inflammatory regulators of IL-6 and TNF-α to activate nucleotide-binding oligomerization domain, leucine rich repeat, and pyrin domaincontaining protein 3 (NLRP3), indirectly regulating the apoptosis of DA neurons[60–63].
For mitochondrial oxidative stress caused by mutation, the generated ROS will further promote the aggregation of α-synuclein, thus aggravating the neuroinflammation induced by α-synuclein[59]. In addition, mutations in ATP13A2 cause impaired iron homeostasis and iron deposition which is linked to the metabolism of glial cells, then the elevated iron concentrations in the brain may affect microglia activation, contributing to the release of inflammatory factors[64–65]. Whether the ATP13A2 mutation will produce the same process of the effects on astrocytes as on microglia has not been reported, but we hypothesize that for the abnormal aggregation of αsynuclein caused by the ATP13A2 mutation, oxidative stress in mitochondria, impaired metal ion homeostasis and metal ion toxicity will also cause astrocytes to produce inflammatory factors. Interestingly, previous studies in our lab have shown that the loss of ATP13A2 function in astrocytes can cause cathepsin B release from lysosomes, leading to the activation of astrocytic NLRP3 inflammasomes, and further aggravating DA neuronal injury[66–67]. The loss of function of mutated ATP13A2 affects glial cells through multiple pathways, prompting the release of inflammatory factors, leading to an inflammatory response in CNS, and further exacerbating the development of PD.
As mentioned earlier, neural cells delivered αsynuclein to cell exteriorsviaexocytosis under ATP13A2 regulation. α-Synuclein was then absorbed and decomposed by astrocytes, a process regulated by ATP13A2. The loss of function of ATP13A2 in astrocytes impairs α-synuclein clearance pathway and leads to activation of NLRP3 inflammasomes in astrocytes, which in turn induces DA neuronal damage. Therefore, the effects of ATP13A2 mutations in PD are not limited to neurons.
ATP13A2 mutation in other neurodegenerative diseases
Studies showed that not only in PD, ATP13A2 mutations were still implicated in hereditary spastic paraplegia (HSP), neurodegeneration with brain iron accumulation (NBIA), and neuronal ceroid lipofuscinosis (NCL) (Table 1). Importantly, there's a lot of overlap between these diseases with PD. HSP is a neurodegenerative disorder characterized by spasms of the lower extremities. Patients with HSP caused by ATP13A2 mutation exhibit psychiatric symptoms,which were the main difference from other associated mutations[68]. In one family with HSP, three siblings were reported to have intellectual disabilities and psychiatric symptoms. Patients presented with hallucinations and delusions prior to the onset of gait disorder, and neurological degeneration was observed[69]. HSP occurs primarily in adults and has been shown to have the same ATP13A2 mutation results as PD, such as a sharp decline in dopamine transport and impaired mitochondrial and lysosomal integrity[33].
In addition, PD mediated by ATP13A2 mutations appeared to induce a type of NBIA, and iron deposits were identified in PD patients with ATP13A2 mutation. As we all know, mitochondria is an important organelle for iron utilization in neurons, and iron plays an important role in energy metabolism[70].ATP13A2 can regulate the stable state of iron, and the mutation of ATP13A2 may induce the destruction of iron homeostasis, which leads to mitochondrial oxidative stress, and ultimately results in the lack of neuronal structure and function[44,70–71].
NCL is also a neurodegenerative disease that mainly occurs in adults and has some similarities with PD. NCL is characterized by the accelerated accumulation of lipofuscin in autophagic vacuoles of neural cells due to lysosomal storage disorders. It has been found that a proliferation of astrocytes and microglia is observed in ATP13A2 knockout mice,with lipid proliferation identified as an important pathological manifestation of NCL. Then, the mice showed motor deficits and strong lipofuscin deposition[72], suggesting that ATP13A2 mutation can lead to NCL.
In a word, ATP13A2 mutations can cause different neurodegenerative diseases. Although with many similarities, the key points of ATP13A2 mutations in different diseases remain unknown. These observations warrant further study and will help identify novel treatments for these neurodegenerative diseases.
Conclusion
Unequivocally, ATP13A2 plays an important role in maintaining physiological function of DA neurons.Mutations in ATP13A2 lead to abnormalities in the metal-cationic, protein, polyamines, and glycolysis processes in neurons, inevitably followed by the dysfunction of ER, mitochondria, and lysosomes, and even the death of DA neurons, exacerbating the development of PD (Fig. 2). Nevertheless, current studies on the role of ATP13A2 in PD pathogenesis have been limited to the regulation of lysosomal function in neurons, and the mechanisms of ATP13A2 regulation of other cell types in the CNS such as oligodendrocytes or other organelles such as mitochondria have not been thoroughly investigated.As illustrating the function of ATP13A2, uncovering its potential mechanism and deepening the understanding of ATP13A2 function will help researchers discover new directions and key molecular targets for PD therapy.

Table 1 The role of ATP13A2 in some of other neurodegenerative diseases
Acknowledgments
The work reported herein was supported by the grants from the National Natural Science Foundation of China (Grant No. 81803505) and Jiangsu Research Hospital Association for Precision Medication (Grant No. JY202134).
 THE JOURNAL OF BIOMEDICAL RESEARCH2022年2期
THE JOURNAL OF BIOMEDICAL RESEARCH2022年2期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Should we open or close the suction port of bronchial blocker during one-lung ventilation?
- Is a designated arterial catheter indicated in transcatheter aortic valve replacement procedure?
- Unilateral pleural effusion secondary to Takayasu arteritis: a case report and literature review
- Valproic acid enhances neurosphere formation in cultured rat embryonic cortical cells through TGFβ1 signaling
- Metabolic syndrome and its association with components of sarcopenia in older community-dwelling Chinese
- Simultaneous determination of clopidogrel, 2-oxo-clopidogrel,and the thiol metabolite of clopidogrel in human plasma by LC-MS/MS