
Theoretical verification of intermolecular hydrogen bond induced thermally activated delayed fluorescence in SOBF-OMe?
2021-12-22 06:44:02MuZhenLi李慕臻FeiYanLi李飛雁QunZhang張群KaiZhang張凱YuZhiSong宋玉志JianZhongFan范建忠ChuanKuiWang王傳奎andLiLiLin藺麗麗
Chinese Physics B 2021年12期
Mu-Zhen Li(李慕臻), Fei-Yan Li(李飛雁), Qun Zhang(張群), Kai Zhang(張凱), Yu-Zhi Song(宋玉志),Jian-Zhong Fan(范建忠), Chuan-Kui Wang(王傳奎), and Li-Li Lin(藺麗麗)
Shandong Key Laboratory of Medical Physics and Image Processing&Shandong Provincial Engineering and Technical Center of Light Manipulations,School of Physics and Electronics,Shandong Normal University,Jinan 250358,China
Keywords: organic light-emitting diodes,thermally activated delayed fluorescence,intermolecular hydrogen bond,decay rates
1. Introduction
Compared with the liquid crystal display(LCD),organic light-emitting diodes(OLEDs)display,which are thinner provide better image quality and contrast.[1]On the basis of the spin-statistics,the ratio of singlet excitons produced in organic electroluminescent materials is one-quarter, and the ratio of triplet excitons is three-quarters.[2,3]For conventional fluorescent OLEDs, the internal quantum efficiency (IQE) is much lower than other OLDEs due to the unavailability of triplet excitons. Since OLEDs with thermally activated delayed fluorescence (TADF) emitters which can obtain luminous singlet and triplet excited states through an effective reverse intersystem crossover (RISC) process were reported to achieve nearly 100%exciton efficiency,TADF materials have attracted widespread attention.[4–6]Although the current application of TADF-OLEDs is still far inferior to phosphorescent OLEDs,due to the high cost and limited resources of phosphorescent materials, TADF-OLEDs still has a lot of room for development as next-generation electroluminescent material.[7–11]For TADF molecules, the most common structure is the donor–acceptor (D–A) or D–A–D configuration. The separation of highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital(LUMO)relies on such twisted,spiral or bulky connection structures, which will effectively reduce the overlap between HOMO and LUMO,and thus reduce the energy gap between S1and T1(?EST).[12–14]There is also TADF produced in the host–guest doped system.[15–17]The characteristics of the host can directly affect the energy of the charge transfer (CT) state and the the ?ESTof the singlet state and the triplet state.[18]As host usually has a large energy gap, the vibration of singlet excitons on the TADF guest are generally well restricted.[19]Recent studies have found that the production of TADF can be assisted by intramolecular or intermolecular hydrogen bonds,[20,21]which indicates that intra-or intermolecular interactions can also promote the formation of TADF,while lack of theoretical verification made it difficult to be understood. Therefore,theoretical studies of the photophysical properties of TADF molecules with intra- or intermolecular hydrogen bonds will contribute to the understanding of the TADF luminescence mechanism. In this paper, we focus on 4-(4-((4-methoxyphenyl)sulfonyl) phenyl) dibenzo[b,d]furan (SOBF-OMe) and 4-(4-(phenylsulfonyl)phenyl)dibenzo[b,d]furan (SOBF-H) (as shown in Fig. 1(a), Fig. S1(a)) reported by Chi’s group.[22]Therein, the TADF is observed for the SOBF-OMe in crystalline state, while no TADF is observed for it in solvent or for SOBF-H in crystalline state. It is deduced that the TADF for SOBF-OMe is generated through intermolecular hydrogen bond. To verify the generation mechanism of TADF in SOBF-OMe, adopting the quantum mechanics and molecular mechanics (QM/MM) method to simulated the lightemitting property of two molecules in crystalline state.[23]Both monomers and dimers are used as models to exploring the origin of TADF.Our results will help to better understand the TADF phenomenon in SOBF-OME crystals, and expand the luminescence mechanism of TADF molecules, thus promoting the development of new multifunctional luminescent TADF molecules.
2. Theoretical methods
In this work, density functional theory (DFT) was used to optimize the ground state (S0). We used the DFT method to optimize the ground state structures of molecules SOBFOMe and SOBF-H.In addition,the excited state properties of TADF molecules directly determine the luminescence properties, the time-dependent density functional theory (TD-DFT)to optimize the excited states. The frequency calculation of the optimized structure confirms the stability of the obtained geometric structure. In order to simulate the solvent of toluene,we used the polarizable continuum model(PCM),which can accurately predict the effect of solvents on TADF molecules.[24–26]The combination of quantum mechanics and molecular mechanics(QM/MM)is used to simulate the properties of molecules in the aggregation state which has been proven reliable.[27–30]Meanwhile,we use a two-layer ONIOM model(as shown in Fig.1(b),and Fig.S1(b))with the center as the high layer and the surrounding as the low layer,where the high layer is calculated using the QM method and the low layer is calculated using the MM method. Further more,we use the universal force field(UFF)and TD-DFT methods for the components of MM and QM,respectively.[31–33]Three functionals(TPSSH, B3LYP, BMK) and two range separation functionals(CAM-B3LYP,ωB97XD)are tested and the 6-31G(d)basis set is used (as listed in Table 1). For dimers, the above functional combined with the empirical dispersion correlation(GD3)is calculated.[34]The calculated emission wavelengths of SOBF-OMe and SOBF-H in the solid phase calculated with the B3LYP functional are found to be similar to the experimental results.Therefore,the subsequent calculations were carried out at the level of B3LYP/6-31G(d). Perform the above calculation process in the Gaussian 16 program.[35]
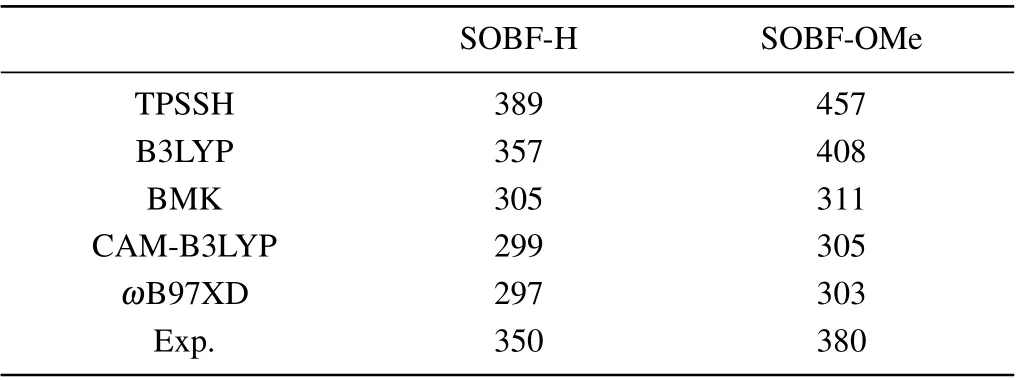
Table 1. Emission wavelengths calculated value with different functionals for SOBF-H and SOBF-OMe in aggregation state(in unit nm).
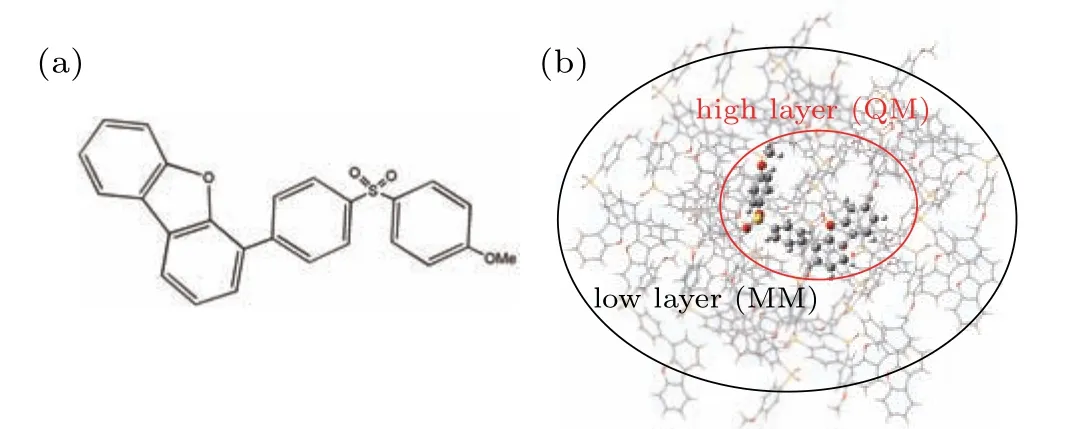
Fig.1. (a)Chemical structures of SOBF-OMe. (b)ONIOM model of SOBFOMe: the center molecule is the high layer,and the others molecules are the low layer.
For the radiative decay rate, the Einstein’s spontaneous emission formula is

whereistands for the atomic ordinal number and ?ρrepresents the gradient vector;abs(?ρ)is that every component of?ρvector takes the absolute value.
Besides, energy decomposition analysis based on the molecular force field is performed to characterize various weak interactions.[41]Most molecular force fields use the form of pair potential to calculate the electrostatic,exchange repulsion and dispersion attraction parts of the non-bonded interaction between atoms.[42]The relevant formula applied is as follows:
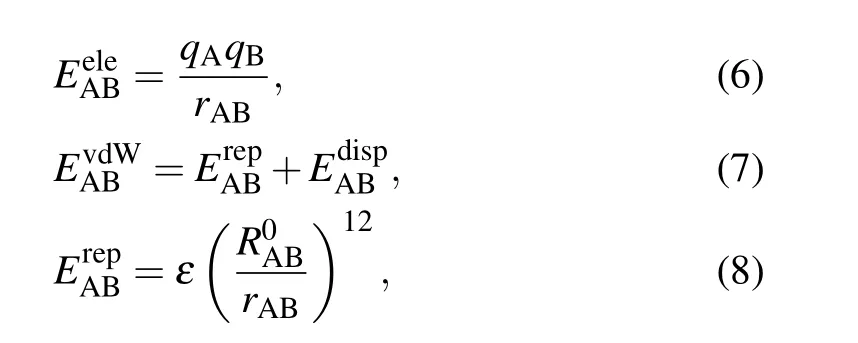
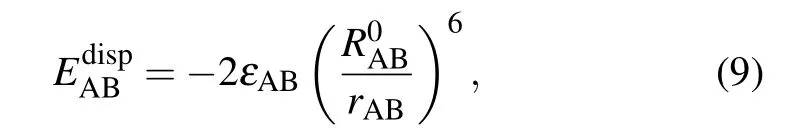
where A and B are the atomic labels,qstands for the atomic charge,ris the distance between atoms,εis the depth of the van der Waals potential well, andR0is the non-bonded distance between atoms.
3. Results and discussion
The light-emitting properties of monomers are studied first to check its possible contribution for TADF.Based on the initial structure in crystal, we optimized the geometry of the molecule in the ground state(S0),the first singlet excited state(S1), and the triplet excited state (TN) in the toluene and aggregation state respectively. At the same time,the root-meansquare displacement (RMSD) method in Multiwfn is combined to show the structural differences between the different states(ground and excited). Moreover,due to different restriction conditions, there is a difference between the optimized molecular geometry in toluene and the optimized molecular geometry in the aggregate state as shown in Fig. 2. It can also be found that there are significant differences between the dibenzofuran and benzene rings. The RMSD values in toluene is much larger than that in aggregation state, indicating that intermolecular interaction could limit the motion of the units in aggregation when they are excited. For TADF molecules,the up-conversion process is closely related to the energy difference between S1and T1(?EST).
Therefore, the adiabatic excitation energies for SOBFOMe in toluene and aggregation state are calculated respectively (see Fig. 3). Further more, the calculated ?ESTin toluene and aggregation state is 1.22 eV and 1.08 eV respectively, indicating that ?ESTcan be reduced by intermolecular interaction. It also indicates that it is difficult for them to realize up-conversion from T1to S1. In addition,the comparison of excited state energy levels in toluene shows that T2,T3,T4,and T5are not only lower than S1but also close to S1in terms of energy. It is suggested that not only T1but also T2, T3,T4,and T5may participate in ISC and RISC processes. In the aggregation state(as shown in Fig.3(b)),we find that the energy of S1is 0.01 eV lower than that of T5. The higher triplet states that close to S1in energy may favor the ISC process,which would be disadvantage to the emission. By comparing the energy structures of molecules in toluene and aggregation state,it is concluded that the interaction between molecules in aggregation state will affect the energy level structure and the ISC and RISC processes of the excited state.
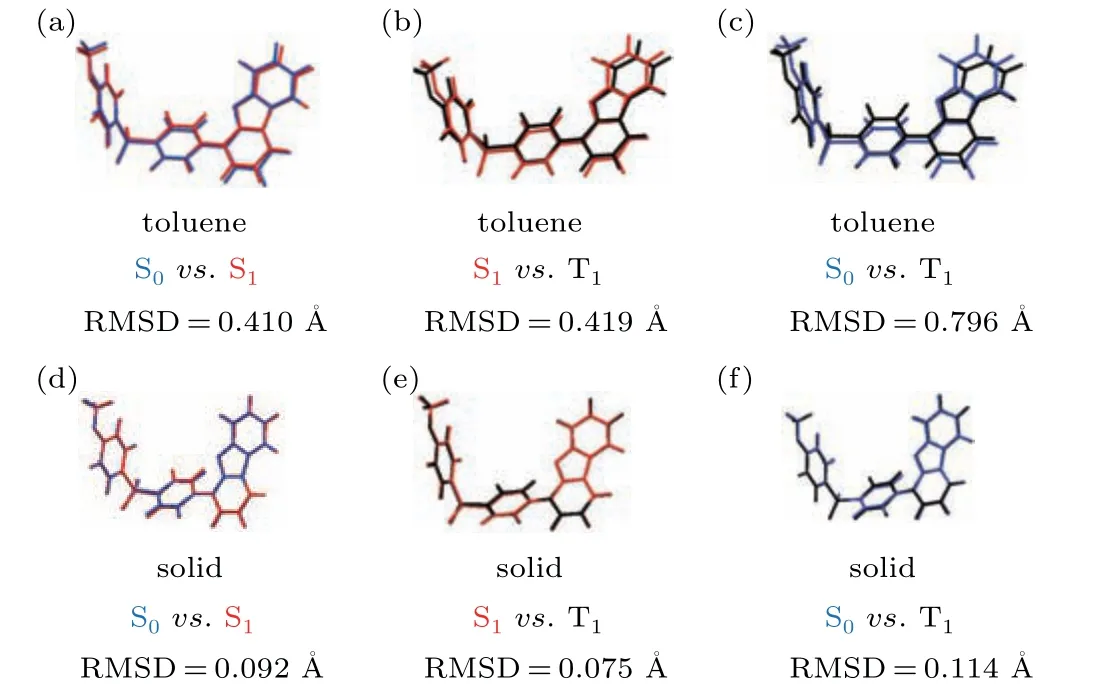
Fig. 2. Geometry difference between S (blue), S1 (red), and T1 (black) in toluene[(a),(b),(c)]and in aggregation state[(d),(e),(f)].

Fig.3. Energy levels for SOBF-OMe in toluene(a)and in aggregation state(b)respectively.
In order to further elucidate the excited state transition properties of SOBF-OMe,we analyzed the natural transition orbital(NTO)and the corresponding transition properties of SOBF-OMe(as shown in Fig.4). Through the transformation of molecular orbital,we qualitatively describe the mechanism of electron excitation.[43,44]The local excited(LE)state(with the LE components are 75%–100%)predicts a large orbital overlap. While the charge transfer(CT)state with strong charge transfer properties(with the LE components are 0%–40%)leads to a small S1–T1energy gap due to significant orbital separation. The hybridized local and charge transfer(HLCT)excited state(with the LE components are 40%–75%)is the LE and CT are mixed,with both LE and CT properties.[45–47]Figures 4(a)and 4(b)show the NTOs of S1and TNobtained by Multiwfn in toluene and aggregated state. We can intuitively see that S1is a typical hybridized local and charge-transfer excited state,there is significant overlap in the conjugation chain between electron and hole,whether it is in toluene or in aggregation state. However,the transitions of the triplet state are mainly distributed in the benzene ring and the dibenzofuran group,except for the T3state. The aggregation state has little effect on the transition properties,compared with that in toluene.

Fig.4. NTOs for S1 and TN sates of SOBF-OMe for monomer in toluene(a)and aggregation state(b)respectively(isovalue is 0.02).
Based on the energy levels of excited states,the ISC processes from S1to TNshould be quite possible in both toluene and aggregation state. The ISC process is not only related to?EST,but also to the spin–orbit coupling(SOC)constant. Table 2 lists the SOC values in toluene and aggregation state,calculated with the quadratic response function method in Dalton package.[48]In aggregation state, the ISC rate between S1and T1(2.03×107s?1) is greater than that in toluene(8.89×105s?1), may be induced by the small SOC constant in toluene. The RISC rates are also calculated based on the SOC values calculated in triplet states. The values are all quite small due to the large energy gap between them. Although the RISC values from T5to S1is comparable to the ISC value,it is meaningless since little excitons would distribute on T5.It proves that it is impossible for the RISC process to happen in the single molecule. Moreover, the radiation decay rate in toluene is 4.43×108s?1, which is larger than that in aggregate state (5.23×107s?1). The main reason should be that the energy difference between S1and S0in aggregate state(3.69 eV) is smaller than that of toluene (3.75 eV). The nonradiation rate in aggregation(1.06×1011s?1)is a little lower than that in toluene(2.92×1011s?1),which may be induced by the confinement of the surroundings.
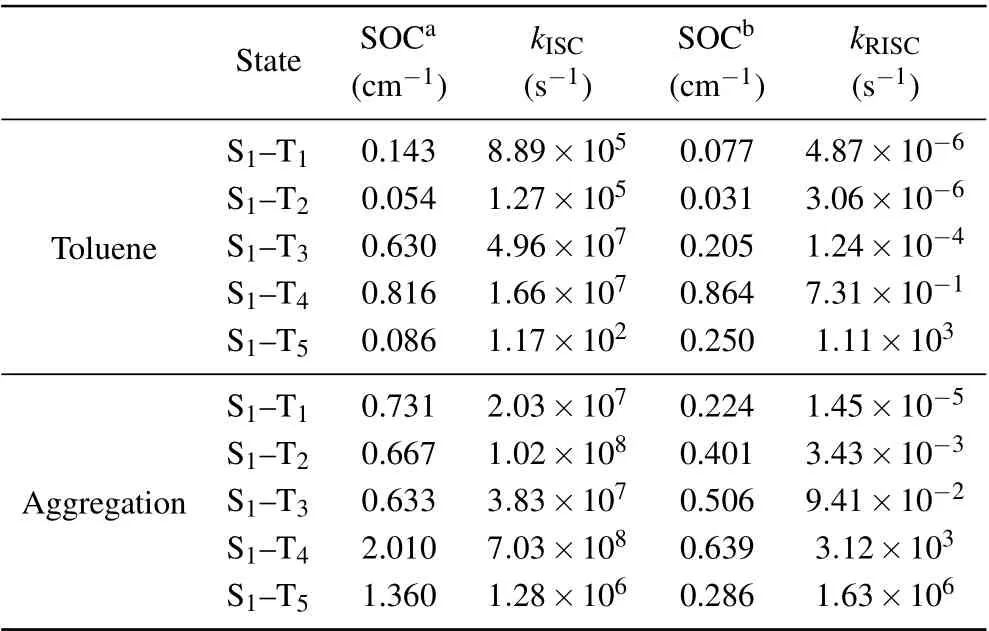
Table 2. Spin–orbit coupling (SOC) constant, intersystem crossing (ISC)rate,and reverse intersystem crossing(RISC)rate between S1 and TN.
The photophysical properties for SOBF-H are also studied in both toluenet and aggregation state. From the energy level of the excited state(see Fig.S2),we find that the excited state energy level of SOBF-H is similar to the SOBF-OMe molecule. In aggregation state, the energy gap between S1and T1(1.18 eV) is slightly larger than that for SOBF-OMe(1.08 eV).The transition orbitals change slightly between the two molecules (see Fig. S3), where the LE component for SOBF-H decreases in S1and increases in T1. In addition,the SOC constant between S1and T5in aggregate state is decreased to (0.368 cm?1) and the ISC rate is increased to(4.30×107s?1) (see Table S1). The RISC rate between S1and T5of SOBF-H(4.44×101s?1)is so small, it is difficult to produce thermally activated delayed fluorescence.

Fig.5. Intermolecular interactions for several dimers described[SOBF-H(a)and SOBF-OMe(b)]through the IGM method.
In experiment, TADF was found for the SOBF-OMe in aggregation. Our calculation results of single molecules indicates that TADF could not be contributed by single molecules.Thus dimers which may contribute to TADF emission are studied. Based on the crystal structure of SOBF-H and SOBFOMe,several dimers are studied,and the interaction between molecules was analyzed by the IGM method (see Fig. 5).Based on the energy decomposition of the AMBER force field, the interaction energy between neighboring molecules is shown in Table 3. For SOBF-OMe it can be seen that the intermolecular interaction of dimer-2 and dimer-4 is weaker,and the intermolecular interaction of dimer-1 is the strongest.Through further analysis, it is found that the dispersion between the two molecules dominates the intermolecular interaction. In addition,there is aπ–πinteraction in dimer-5,it is mainly theπ–πinteraction between dibenzofuran groups. In the same way,π–πinteraction also exists in dimer-3,and theπ–πstacking exists in the donor benzene ring. For SOBF-H the intermolecular interaction of dimer-1 and dimer-2 is weak.Through further analysis,we find that the dispersion between the two molecules dominates the intermolecular interaction.Using IGM analysis,it is found that SOBF-OMe dimer-2 and dimer-4 have obvious intermolecular hydrogen bond interactions.It is also found that dimer-3 and dimer-5 have significantπ–πinterraction. Then several pairs of dimers for SOBF-H and SOBF-OMe are optimized with the QM/MM model. The emission wavelengths and the S1–T1energy gap for all the dimers are shown in Table 4. It can be seen that the calculated emission wavelengths of dimer-2 and dimer-4 for SOBF-OMe consistent with the experimental results of TADF.In addition,the ?ESTof S1and T1in dimer-2 is also much smaller than other dimers, which also predicts the possible up-conversion from triplest states to S1. The NTOs of SOBF-H and SOBFOMe dimer are also analyzed (see Fig. S4). It is found that both two dimers of SOBF-H have delocalized NTOs with electron distributed on both two molecules and significant overlap can be found for transition orbitals. However, the NTOs for dimers except for dimer-5 of SOBF-OMe indicate that electrons transit from one molecule to the other when they are excited. Although dimer-5 has strongπ–πinteraction, it is meaningless to the TADF emission. It favors the emission of phosphorescence.For the energy diagram of excited states,we can find that there are several states lying between S1and T1,which may favor the ISC process and the emission of phosphorescence(see Figs.S5 and S6). The calculated wavelength of T1for all the dimers is also in consistent with the experimental result,which further illustrates that dimers except those with H-bond may favor the phosphorescence emission(see Table S2). Therefore,we believe that the intermolecular H-bond could induce smaller S1–T1energy gap,thus the TADF could be generated. Dimers withπ–πinteraction would favor the phosphorescence emission.
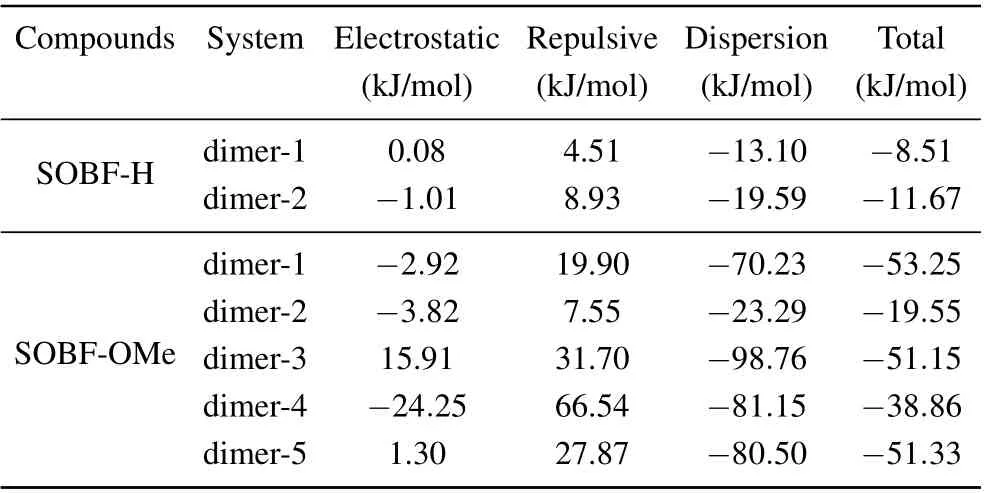
Table 3. Intermolecular interaction energy including electronic, repulsion,and dispersion interactions in several dimers studied.
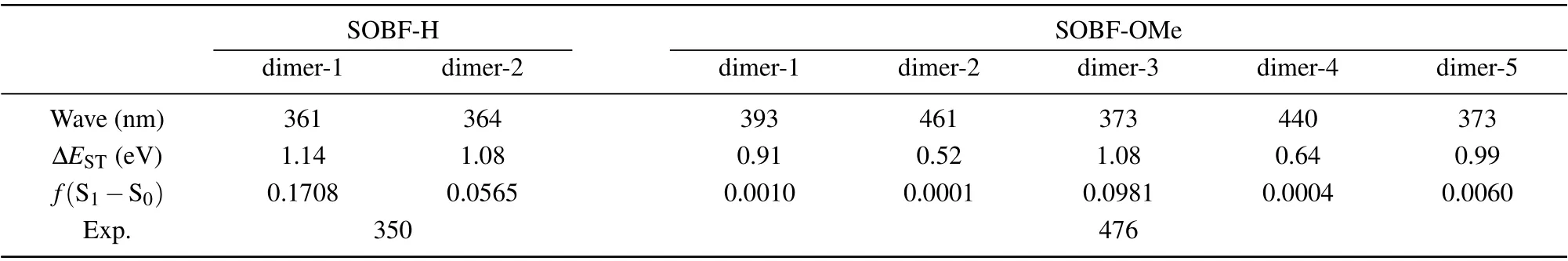
Table 4.Calculated fluorescence wavelength,?EST and oscillator strength of SOBF-OMe and SOBF-H for dimer based on their single crystal structures using TD-DFT method.
4. Conclusion
The excited state properties and decay rates of SOBFOMe in toluene and aggregation state are studied respectively.The results show that the intermolecular interaction could limit the geometric change of molecules when they are excited in aggregation state. The energy gaps between S1and T1are reduced to some extent, while the transition properties have little changed. Theoretical simulation of the single molecular emission in toluene and in aggregation state agrees well the prompt fluorescence and phosphorescence. It is also indicated that TADF could not be generated by single SOBF-OMe molecules but dimers with intermolecular H-bond in crystal.In addition, emission properties of SOBF-H are also investigated for comparison,which shows that dimers without intermolecular H-bond are unable to generate TADF.Our calculation results confirms experimental results that TADF could be induced by dimers with intermolecular H-bond,although that single molecules with large S1–T1energy gap have no contribution to it. The results would favor the development of new type light-emitting molecules with TADF emission.
猜你喜歡
作文周刊·小學(xué)二年級版(2022年44期)2022-11-29 09:32:42
早期教育(家庭教育)(2022年12期)2022-05-30 10:48:04
軟件(2020年3期)2020-04-20 01:44:52
幼兒教育·父母孩子版(2020年8期)2020-03-04 11:54:24
東坡赤壁詩詞(2018年4期)2018-11-07 11:01:44
水動力學(xué)研究與進展 B輯(2018年3期)2018-07-06 10:02:00
東坡赤壁詩詞(2018年2期)2018-05-10 11:08:24
語文教學(xué)與研究(綜合天地)(2017年1期)2017-02-17 20:25:11
永善文學(xué)(2016年4期)2016-11-19 09:41:08
小天使·四年級語數(shù)英綜合(2016年10期)2016-10-12 08:54:49
- Chinese Physics B的其它文章
- Modeling the dynamics of firms’technological impact?
- Sensitivity to external optical feedback of circular-side hexagonal resonator microcavity laser?
- Controlling chaos and supressing chimeras in a fractional-order discrete phase-locked loop using impulse control?
- Proton loss of inner radiation belt during geomagnetic storm of 2018 based on CSES satellite observation?
- Embedding any desired number of coexisting attractors in memristive system?
- Thermal and mechanical properties and micro-mechanism of SiO2/epoxy nanodielectrics?