
The high diversity of SARS-CoV-2-related coronaviruses in pangolins alerts potential ecological risks
2021-02-10 13:07:30MinShengPengJianBoLiZhengFeiCaiHangLiuXiaoluTangRuochenYingJiaNanZhangJiaJunTaoTingTingYinTaoZhangJingYangHuRuNianWuZhongYinZhouZhiGangZhangLiYuYongGangYaoZhengLiShiXueMeiLuJianLuYaPingZhang
Zoological Research 2021年6期
Min-Sheng Peng, Jian-Bo Li, Zheng-Fei Cai, Hang Liu, Xiaolu Tang, Ruochen Ying, Jia-Nan Zhang,Jia-Jun Tao, Ting-Ting Yin, Tao Zhang, Jing-Yang Hu, Ru-Nian Wu, Zhong-Yin Zhou, Zhi-Gang Zhang, Li Yu,Yong-Gang Yao, Zheng-Li Shi, Xue-Mei Lu,2,9, Jian Lu,*, Ya-Ping Zhang,2,3,,9,*
1 State Key Laboratory of Genetic Resources and Evolution, Yunnan Laboratory of Molecular Biology of Domestic Animals, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650201, China
2 Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, Yunnan 650204, China
3 KIZ/CUHK Joint Laboratory of Bioresources and Molecular Research in Common Diseases, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, Yunnan 650201, China
4 State Key Laboratory for Conservation and Utilization of Bio-resources in Yunnan, Yunnan University, Kunming, Yunnan 650091, China
5 State Key Laboratory of Protein and Plant Gene Research, Center for Bioinformatics, School of Life Sciences, Peking University, Beijing 100871, China
6 Molbreeding Biotechnology Co., Ltd., Shijiazhuang, Hebei 050035, China
7 Key Laboratory of Animal Models and Human Disease Mechanisms of the Chinese Academy of Sciences & Yunnan Province, Kunming Institute of Zoology, Kunming, Yunnan 650201, China
8 CAS Key Laboratory of Special Pathogens and Biosafety, Wuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, Hubei 430071, China
9 Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, Yunnan 650201, China
ABSTRACT
Understanding the zoonotic origin and evolution history of SARS-CoV-2 will provide critical insights for alerting and preventing future outbreaks.A significant gap remains for the possible role of pangolins as a reservoir of SARS-CoV-2 related coronaviruses (SC2r-CoVs).Here, we screened SC2r-CoVs in 172 samples from 163 pangolin individuals of four species, and detected positive signals in muscles of four Manis javanica and, for the first time, one M.pentadactyla.Phylogeographic analysis of pangolin mitochondrial DNA traced their origins from Southeast Asia.Using in-solution hybridization capture sequencing, we assembled a partial pangolin SC2r-CoV (pangolin-CoV) genome sequence of 22 895 bp (MP20) from the M.pentadactyla sample.Phylogenetic analyses revealed MP20 was very closely related to pangolin-CoVs that were identified in M.javanica seized by Guangxi Customs.A genetic contribution of bat coronavirus to pangolin-CoVs via recombination was indicated.Our analysis revealed that the genetic diversity of pangolin-CoVs is substantially higher than previously anticipated.Given the potential infectivity of pangolin-CoVs, the high genetic diversity of pangolin-CoVs alerts the ecological risk of zoonotic evolution and transmission of pathogenic SC2r-CoVs.
Keywords: SARS-CoV-2 ; Pangolin ; Recombination ;Diversity; Sequencing; mtDNA
INTRODUCTION
The COVID-19 pandemic, caused by the global spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has had significant impacts on the global economy and public health (Hu et al., 2021).Efforts to trace the zoonotic origin of SARS-CoV-2 and investigate its evolution and transmission have attracted worldwide attention (Banerjee et al., 2021; Hu et al., 2021; WHO, 2021; Wong et al., 2020;Zhang & Holmes 2020).Recent studies have identified coronaviruses closely related to SARS-CoV-2 (genomic identity >90%) in various bat species, includingRhinolophus affinis(Zhou et al., 2020b),R.malayanus(Zhou et al., 2020a),R.cornutus(Murakami et al., 2020),R.shameli(Hul et al.,2021),R.acuminatus(Wacharapluesadee et al., 2021), andR.pusillus(Zhou et al., 2021).Among these bat-derived SARS-CoV-2-related coronaviruses (SC2r-CoVs), RaTG13 fromR.affinisis the closest to SARS-CoV-2, with the sequence identity of 96.2% between the two viral genomes(Zhou et al., 2020b).
Besides bats, SC2r-CoVs have also been detected in the Malayan pangolins (Manisjavanica) (Lam et al., 2020; Liu et al., 2020; Xiao et al., 2020; Zhang et al., 2020).The pangolinderived SC2r-CoVs (pangolin-CoVs) can be classified into two sublineages.The first sublineage, defined by a pangolin-CoV strain detected in Malayan pangolins confiscated during antismuggling operations by Guangdong Customs (pangolin-CoVGDC), shares a genomic sequence similarity of 92.4% to SARS-CoV-2 (Liu et al., 2020; Xiao et al., 2020; Zhang et al.,2020).The second sublineage was defined with six strains of pangolin-CoVs that were identified in Malayan pangolins obtained during anti-smuggling operations performed by Guangxi Customs (pangolin-CoV-GXC) (Lam et al., 2020).The genome sequences of the six pangolin-CoV-GXC strains were highly similar to each other (>99%), and they overall show a similarity of 85.5% to that of SARS-CoV-2 (Lam et al.,2020).The two pangolin-CoV sublineages share a genomic sequence similarly of 84%, presenting substantial genetic divergence.Currently,M.javanicais the only known pangolin species that harbors coronaviruses closely related to SARSCoV-2.Although the pangolin-CoVs tend to be more distantly related to SARS-CoV-2 than the aforementioned bat-derived SC2r-CoVs, recent studies show that the receptor-binding domain (RBD) of both pangolin-CoV sublineages can potentially bind the human ACE2 receptor (Dicken et al., 2021;Guo et al., 2021; Nie et al., 2021; Niu et al., 2021; Zhang et al., 2021), raising concerns about the potential risks of spillover of these pangolin-CoVs to humans.
Different hypotheses have been proposed to account for the possible role of pangolins as a source of potential zoonosis of SC2r-CoVs (see Supplementary Table S1 for a summary).Based on the studies that the two divergent pangolin-CoV sublineages were detected in the samples ofM.javanicacollected before the COIVD-19 pandemic, one hypothesis is that pangolins are reservoirs or natural hosts of SC2r-CoVs(the natural host hypothesis).According to this hypothesis,one or both sublineages of pangolin-CoVs might have a long history of circulating in pangolins in the wild (Lam et al., 2020;Liu et al., 2020; Zhang et al., 2020).However, a recent study in which 334 liveM.javanicaindividuals confiscated in Peninsular Malaysia and Borneo were screened for pangolin-CoVs yielded no positive results, suggesting that pangolins are only incidental hosts of SC2r-CoVs in smuggling networks,and that SC2r-CoVs were recently transmitted from bats or other unknown animals to pangolins (the incidental host hypothesis) (Lee et al., 2020).Indeed, unlike bats, which typically carry coronaviruses asymptomatically, some pangolin individuals infected with SC2r-CoVs have shown clinical signs of severe respiratory disease (Lam et al., 2020; Liu et al.,2019, 2020; Xiao et al., 2020); this is in line with the incidental host hypothesis.If this hypothesis is correct, the presence of SC2r-CoVs with high similarity to pangolin-CoVs would be expected in wildlife across the same region in which pangolins are distributed or along pangolin smuggling routes.However,no evidence of such SC2r-CoVs has yet been found despite extensive surveys of wildlife and livestock in China and Southeast Asia (Hul et al., 2021; Wacharapluesadee et al.,2021; WHO, 2021).Collectively, there is yet no consensus view on the two competing hypotheses, and more studies are required for a better understanding of whether pangolins act as natural, incidental, or intermediate hosts.
Here, we first screened the SC2r-CoVs in 172 samples from 163 pangolin individuals belonging to four species that were confiscated during anti-smuggling operations in Yunnan Province of southwestern China.Then we analyzed the genome sequence of a pangolin-CoV that was isolated and sequenced from aM.pentadactylasample (MP20).Our results, together with the re-analyses of pangolin-CoVs sequenced in a previous study (Lam et al., 2020), reveal that the genetic diversity of pangolin-CoVs are substantially higher than previously anticipated, and provide more evidence that favors the natural host over the incidental host hypothesis.
MATERIALS AND METHODS
Sample collection
This study was approved by the Zoology Animal Care and Ethics Committee of the Kunming Institute (SMKX-20 200 210-01).The pangolin samples were collected by the Animal Branch of the Germplasm Bank of Wild Species of the Chinese Academy of Sciences from seizures of illegal wildlife trade and smuggling in the international border regions of Yunnan Province, southwestern China, between 1990 and 2018.All the pangolin individuals had been dead when the samples were collected, with the postmortem intervals unknown.The samples were collected and preserved in 95% alcohol at –80 °C immediately after sample collection.
Viral RNA extraction
The samples were disrupted in 200 μL of PBS by ultrasonication for 5 min.Viral RNA was extracted using the High Pure Viral RNA Kit (Roche, Switzerland) according to the manufacturer-recommended procedure.After elution with 50 μL of RNase-free water, the RNA purity (OD260/280) and concentration and the absorption peak were measured with a NanoDrop 2000 (Thermo Scientific, USA).
RT-PCR and Sanger sequencing
We followed a published RT-PCR strategy (Wu et al., 2020) to screen the viral RNA extracts.PCR primers and a 5'FAM-labeled probe (F1: 5’-GGTCATGTGTGGCGGCTC-3’; R1: 5’-GCTGTAACAGCTTGACAAATGTTAAAG-3’; Probe: 5’-CTATATGTTAAACCAGGTGGAAC-3’) were designed to target theSgene of SARS-CoV-2.AgPath-ID? One-Step RTPCR reagents (Thermo Scientific, USA) were used for amplification in a QuantStudio 12K Flex Real-Time PCR System (ABI, USA) as follows: 50 ℃ for 10 min, 95 ℃ for 10 min, and 40 cycles consisting of 95 ℃ for 15 s and 55 ℃ for 45 s, followed by a default melting curve step.Each run was repeated at least three times.The PCR products from RTPCR-positive samples were TA cloned with the vector pMD?18-T (Takara, Japan) and subsequently Sanger sequenced.We then aligned the sequences using BLAST with the 2019 Novel Coronavirus Resource (2019nCoVR) (Gong et al., 2020).
Library preparation and screening
Viral RNA sequencing library preparation and depletion of ribosomal RNA (rRNA) were performed using the SMARTer Stranded Total RNA-seq Kit V2-Pico Input Mammalian(Takara, Japan) according to the manufacturer’s protocol, with 400 ng of input RNA.The five libraries were PE150 sequenced on the NovaSeq 6000 platform (Illumina, USA) by Novogene Co., Ltd., China.
In-solution capture and next-generation sequencing
We used an in-solution capture kit and corresponding protocol that was originally developed for laboratory diagnosis of SARS-CoV-2 by Molbreeding Biotechnology Co., Ltd., China.A total of 502 GenoBaits DNA probes with an average length of 120 nucleotides were designed based on 50 different human SARS-CoV-2 genome sequences.A similar system was applied to capture and enrich SARS-CoV-2 viral sequences from viral particles at low concentrations (Wen et al., 2020).The coverage depth for the regions with mutations across diverse viral lineages ranged from 2 to 3 X to enhance the capture capacity (Johnson et al., 2019).A total of 500 ng of RNAs was used for in-solution capture.The commercial kit,together with the protocol, is available from Molbreeding Biotechnology Co., Ltd, China.Finally, the GenoBaits DNA probe capture library was PE150 sequenced on the HiSeq X Ten platform (Illumina, USA) by Berry Genomics Co., Ltd.,China.
Analysis of NGS data
Reads that did not include one of the tags/indices used in library construction were discarded to avoid potential contamination (Briggs et al., 2007; Green et al., 2009).We removed the tag/index and adaptor sequences with Cutadapt v2.10 (Martin, 2011).Reads with a length less than 25 bp were excluded.We used BWA-MEM 0.7.15-r1140 (Li, 2014)to map the reads to the 21 coronaviral genomes(Supplementary Table S2) with the parameter -M -k 15.SAMtools v1.9 was used to filter unmapped reads with the parameter -F 4 (Li et al., 2009).Duplicates were marked by using the MarkDuplicates module of GATK v4.1.3.0 (McKenna et al., 2010) with default parameters.Duplicate reads were removed by SAMtools with the parameter -F 1024.
Genome assembly
We downloaded 21 representative coronavirus genomes(Drexler et al., 2010; Ge et al., 2013; He et al., 2014; Hu et al.,2018; Hul et al., 2021; Lam et al., 2020; Lau et al., 2005; Li et al., 2005, 2021; Lin et al., 2017; Liu et al., 2020; Murakami et al., 2020; Qin et al., 2003; Tao & Tong, 2019;Wacharapluesadee et al., 2021; Wu et al., 2020; Zhou et al.,2020a, 2020b) presenting relatively high similarities to SARSCoV-2 especially when we conducted the analysis.We used MP789 and GXP5L as representatives of the pangolin-CoVGDC (MP789) and pangolin-CoV-GXC (GXP5L) substrains,respectively.We performeddenovoassembly of the 21 coronavirus-mapped reads with SPAdes v3.15.2 (Bankevich et al., 2012) and MegaHit v1.2.9 (Li et al., 2015).We queried the assembled contigs by using BLAST to search NCBI nucleotide collection standard databases to exclude contigs unrelated to coronaviruses.Reference-guided scaffolding was conducted to align the contigs and reads mapped to the 21 coronaviruses that were not assembled into contigs to the reference genome of pangolin-CoV-GXC (GXP5L) (Lam et al.,2020) with blastn v2.10.0+.Reads that mapped to the 21 coronavirus sequences were remapped to the consensus sequence by using BWA-MEM (Li, 2014) and checked by IGV(Robinson et al., 2011).The gaps in the assembled consensus sequence were filled in with “N” to facilitate subsequent multiple genome sequence alignment.The sequencing coverage depth was estimated with SAMtools (Li et al., 2009).
Phylogenetic and evolutionary analysis
We conducted a phylogenetic analysis of MP20 in the context of 21 coronavirus genomes (Supplementary Table S2).A total of 21 sequences were aligned with the assembled consensus sequence with the online service MAFFT v7 (Katoh et al.,2019).The alignment was checked manually with reference to the genome annotation of MN908947.The substitution models and related parameters were determined by the Bayesian information criterion (Schwarz, 1978) in ModelTest-NG v0.1.6(Darriba et al., 2020).Nucleotide substitution saturation was tested using DAMBE7 (Xia, 2018).MatGAT v2.01 was used to analyze sequence similarity and identity (Campanella et al.,2003).The results were visualized with SimPlot v3.5.1 (Lole et al., 1999).All gaps and indels were discarded.The maximum likelihood phylogenetic trees for nucleotide alignments were constructed on the basis of 1 000 bootstrap replications by using MEGA X v10.1 (Kumar et al., 2018).
The pairwise nucleotide distance based on transitions and transversions was calculated with the Jukes-Cantor model with gamma distribution (G) +4 in MEGA X v10.1 (Kumar et al., 2018).We calculated the nonsynonymous/synonymous rate ratio (dN/dS) between (1) pangolin-CoV-GDC (MP789)and pangolin-CoV-GXC (GXP5L) and between (2) pangolin-CoV-GXC (GXP5L) and MP20 with the program YN00 implemented in PAML v4.9a (Yang, 2007).
Intra-host variant analysis
Six pangolin-CoV sample sequences were downloaded from the Sequence Read Archive (accession numbers:SRR11093266, SRR11093267, SRR11093268,SRR11093269, SRR11093270, and SRR11093271).The reads were mapped to the pangolin-CoV-GXC reference genome (GXP5L) using BWA-MEM (Li, 2014).SAMtools v1.9 was used to filter out reads with a mapping quality less than 30.Freebayes v9.9.2-27-g5d5b8ac (Garrison & Marth, 2012)was used to call the iHVs (--min-mapping-quality 20 --minbase-quality 20).The GXP3B library (SRR11093270) was excluded due to low coverage depth.
Recombination analysis
We used RDP, GENECONV, BootScan, MaxChi, Chimaera,SiScan and 3Seq implemented in RDP v.4.101 to detect putative recombination events (Martin et al., 2015).SimPlot v.3.5.1 was employed to visualize the recombination events and breakpoints in a sliding window (Lole et al., 1999).The candidate recombination regions were further checked by constructing neighbor-joining trees with the Kimura 2-parameter model and 1 000 bootstrap replications by using MEGA X v10.1 (Kumar et al., 2018).
Pangolin mitochondrial DNA analysis
The DNA barcoding classification of four pangolin samples(MP20, MJ20, MJ57, and MJ54) was published previously (Hu et al., 2020).An 829 bp region of the mitochondrialCOIgene and a 790 bp region of the cytbgene were sequenced for MJ18007 according to published protocols (Hu et al., 2020;Nash et al., 2018).TheCOIsequence was queried using BLAST searches of GenBank to identify the pangolin species.Mitochondrial DNA clustering based on theCOIand cytbgenes was performed according to the protocol used in a reported phylogeographic analysis of pangolins (Hu et al.,2020).M.pentadactylaMP20 was classified into the MPBmtcluster, indicating that it likely originated in Myanmar (Hu et al., 2020).M.javanicaMJ20 was assigned to the MJAmtcluster, restricted it to northern Mainland Southeast Asia (e.g.,Myanmar), while MJ54, MJ57, and MJ18007 were classified into the MJBmtcluster, which is prevalent in Island Southeast Asia (Hu et al., 2020).In particular, MJ18007 shared its mtDNA haplotype with the sample MZBR_1 085, recorded as seized illegal trade from Borneo in GenBank (COI:MG825608; cytb: MG825549) (Nash et al., 2018).
RESULTS
Screening SC2r-CoVs in samples of four pangolin species
To gain a more comprehensive view on the prevalence of SC2r-CoVs in pangolins, we performed reverse-transcription PCR (RT-PCR) of viral RNA extracted from muscles of 172 samples collected from 163 pangolin individuals that were confiscated during anti-smuggling operations in Yunnan Province, southwestern China, between 1990 and 2018.To infer the species and potential geographic sources of the pangolins, we analyzed mitochondrial DNA using a pangolin phylogeographic framework as described previously (Hu et al.,2020).Altogether, in this study, we tested 172 samples (163 muscles, two blood samples, three kidneys, two small intestines, one heart, and one liver) from 163 pangolin individuals of 4 species (M.javanica, 106;M.pentadactyla,53;Phataginustricuspis, 3; andP.tetradactyla, 1).
We followed a published RT-PCR strategy (Wu et al., 2020)that target a 121-nucleotide fragment of theSgene of SARSCoV-2 to screen the viral RNA extracts.In total, we detected positive RT-PCR signals in muscles of fourM.javanicasamples and, for the first time, oneM.pentadactylasample(Figure 1A).Sanger sequencing revealed that the RT-PCR fragment was 81.01%–83.54% similar to the SARS-CoV-2 sequence (MN908947).Phylogeographic analysis of mitochondrial DNA showed that the five pangolin individuals were obtained from a wide area across Southeast Asia(Figure 1A).
Low amounts of viral RNA in pangolin samples
To determine the genome sequences of the SC2r-CoVs from the five muscle samples that yielded positive RT-PCR results,we performed viral RNA sequencing for each sample with the Illumina platform (each mRNA-Seq library generated 16.6–18.4 Gb of data).The majority (93.44%–97.25%) of the mRNA-Seq reads were mapped to the pangolin reference genomes (Hu et al., 2020).Only twelve reads from sample MP20 (M.pentadactyla) and eight reads from sample MJ57(M.javanica) could be mapped to one of the 21 known SC2r-CoVs and SARS-related CoVs (SARSr-CoV) genomes that were discovered in bats and pangolins in previous studies(Tables S2, S3), and we failed to detect reads that can be reliably mapped on these SC2r-CoV and SARSr-CoV genomes in the remaining three libraries.The scarcity of the mapped reads was not unexpected since the pangolin muscle samples were collected after the animals were dead(postmortem intervals unknown), and the samples had been preserved in 95% alcohol, mainly for DNA studies, likely leading to extremely low abundance and high degradation of the RNAs.
A novel partial pangolin-CoV genome sequence (MP20)from M.pentadactyla
The development of targeted enrichment technologies for samples that are degraded has considerably facilitated the sequencing of ancient pathogen genomes (Spyrou et al.,2019).Here, we applied a similar strategy to RNAs extracted from the muscle of theM.pentadactylaindividual (sample MP20), as it showed the highest abundance of SC2r-CoV nucleotides among the five pangolins for which mRNA-Seq libraries were available.Specifically, we carried out in-solution hybridization capture with dense probes that cover the whole genome of SARS-CoV-2 to enrich SC2r-CoV sequences.Targeted enrichment sequencing generated ~74 Gb of data,with 395 424 769 PE150 reads.After a series of quality control checks and data filtering, we obtained 901 clean reads that could be mapped to the 21 known SC2r-CoV and SARSr-CoV genomes (Supplementary Table S2; aligned length >100 nucleotides and blast E<10-5were required).Wedenovoassembled these mapped reads using both SPAdes(Bankevich et al., 2012) and MegaHit (Li et al., 2015).The SPAdes analysis yielded 36 contigs that ranged from 230 to 1 994 nucleotides in length, with a mean length of 525.6 nucleotides.The MegaHit analysis similarly yielded 36 contigs,ranging from 201 to 1 695 nucleotides in length with a mean length of 475.25 nucleotides.Since these contigs were more similar to the pangolin-CoV-GXC sequence than to the other SC2r-CoV sequences we analyzed, we pooled the contigs assembled by SPAdes and MegaHit together, and scaffolded the contigs and some mapped reads (without assembly into contigs), using the GXP5L strain of pangolin-CoV-GXC as a reference.In addition, two unique reads from MJ57 were used to fill two gaps (23 266?23 301 and 25 577?25 588).We finally obtained a consensus genome from sample MP20 that contained sequenced nucleotides at 22 895 sites, and this viral genome (named MP20) covers ~76.6% of the SARS-CoV-2 genome and has a coverage depth from 1 to 64 X and a mean depth of 8.45 X (Figure 1B).
Divergence between MP20 and other pangolin-CoVs
The MP20 genome exhibited 86.3% nucleotide sequence identity and 93.9% amino acid identity to the reference genome of SARS-CoV-2 (MN908947) and the corresponding translated polypeptide, respectively (Figure 1C).The alignment for MP20 (red bar) to SARS-CoV-2 genes indicated 79.47% coverage ofORF1ab, 66.09% ofS, 78.26% ofORF3a, 7.89% ofE, 85.80% ofM, 100% ofORF6, 88.80% ofORF7a, 0.82% ofORF8, 99.52% ofNand 100% ofORF10.MP20 was more similar to pangolin-CoV-GXC (96% sequence identity) than to pangolin-CoV-GDC (86.4% sequence identity).Indeed, the construction of a phylogenetic tree based on the whole-genome sequences of MP20 and the 21 known SC2r-CoV and SARSr-CoVs clustered MP20 with GXP5L (the reference sequence of pangolin-CoV-GXC) in the pangolin-CoV-GXC sublineage (Figure 1D).
Although we did not retrieve the full-length sequence of theSgene of MP20, which encodes the spike protein and plays a crucial role in the binding to the host’s cellular receptor and cellular entry, we achieved an alignment with 66.09% coverage (2 526 bp), mainly covering the S2 subunit(Figure 1C).MP20 and GXP5L showed 95% nucleotide sequence identity and 98.8% amino acid identity at theSgene and spike protein, respectively.There are six amino acids in the RBD region of the S protein that are crucial for the attachment of SARS-CoV-2 to the ACE2 receptor (Lu et al.,2020; Ou et al., 2020; Qu et al., 2005; Ren et al., 2008; Wan et al., 2020; Wrapp et al., 2020), but unfortunately, the MP20 sequence only covers 60.99% of the RBD region and does not cover any of those crucial sites, which hampered further investigation of the potential infection ability of MP20(Supplementary Figure S1).However, our sequence comparisons revealed there were five amino acid changes in the RBD regions between GXP5L and MP20 (I324T, N504D,G520A, T532Q, and N536D; Supplementary Figure S1).Further studies are required to investigate whether those differences might be associated with the difference in the cellular entry between MP20 and GXP5L.
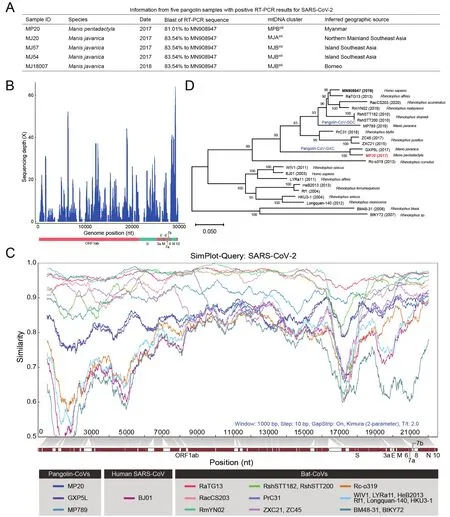
Figure 1 Genetic characterization of pangolin-CoVs in pangolin muscles
The average genome-level nucleotide divergence between MP20 and GXP5L was 0.041, which is very similar to those between human SARS-CoV-2 and the bat SC2r-CoVs RaTG13 (0.036) and RmYN02 (0.045), suggesting the genetic diversity within the pangolin-CoV-GXC sublineage should not be neglected.Previous evolutionary analysis suggested that strong purifying selection underlay the protein changes between SARS-CoV-2 and RaTG13 (Li et al., 2020; Tang et al., 2020).Likewise, we identified strong signals indicating purifying selection between MP20 and GXP5L (genomic average dN/dS=0.011/0.181=0.058; Supplementary Table S4)and between GXP5L and pangolin-CoV-GDC(dN/dS=0.060/0.718=0.083; Supplementary Table S5),suggesting functional constraints have operated the protein evolution of the pangolin-CoV strains.By assuming a molecular clock and a neutral substitutional rate of 1.69×10-3substitutions/site/year (Boni et al., 2020), we estimated the divergence time between pangolin-CoV-GDC and pangolin-CoV-GXC based on synonymous substitutions (dS) as 212.4(0.718/(2×1.69×10-3)≈212.4) years ago and that between GXP5L and MP20 as 53.6 (0.181/(2×1.69×10-3)≈53.6) years ago.Overall, our analyses suggest the divergence of MP20 from other pangolin-CoV strains was not very recent and support the scenarios that these pangolin-CoVs have had a long history of circulating in different species of pangolins in the wild.
Pervasive intra-host variants in the pangolin-CoVs
Intra-host variants (iHV) provide key insights into the genetic diversity and evolutionary dynamics of SARS-CoV-2 (Armero et al., 2021; Wang et al., 2021).Despite the low sequencing coverage of the MP20 coronavirus, we still observed a modest number of intra-host variants (iHVs) at six sites (three of them were nonsynonymous: G1299T and G6105T inORF1aband A26530C inM) that had at least 5X coverage depth in the MP20 sequencing results (Supplementary Table S6).To further examine the occurrence of iHVs in pangolin-CoVs, we analyzed the libraries from six previously published pangolin-CoV-GXC viruses that were detected in various tissues ofM.javanica(Lam et al., 2020), using GXP5L as the reference genome.To avoid sequencing errors, we considered only those sites for both single nucleotide variants and small indels that had coverage of ≥150X and ≥10 reads covering the alternative alleles in each library.The number of identified iHVs per library ranged from 71 to 432 (the GXP3B library SRR11093270 was excluded because it had a median coverage of 5X, and no iHV site met our criteria; Table 1).
In total, we identified 852 iHVs in at least one of five libraries, including 217 synonymous, 336 nonsynonymous,264 frameshift, and 3 stop-gain variants.566 (66.4%) of the iHVs were restricted to a single library (Figure 2A).Consistent with previous observations that intra-host variants of SARSCoV-2 could be shared among samples collected from different patients (Armero et al., 2021; Wang et al., 2021),here, we found a total of 286 (33.6%) of these iHVs were detected in at least two of the five libraries (Figure 2).A total of 158 iHVs were observed in theSgene (Table 1), and 58(36.7%) of theseSgene iHVs were shared by at least twolibraries (Figure 2B).Collectively, the large numbers of iHVs we observed here adds another layer of complexity for interrogating the genetic diversity of the pangolin-CoVs.
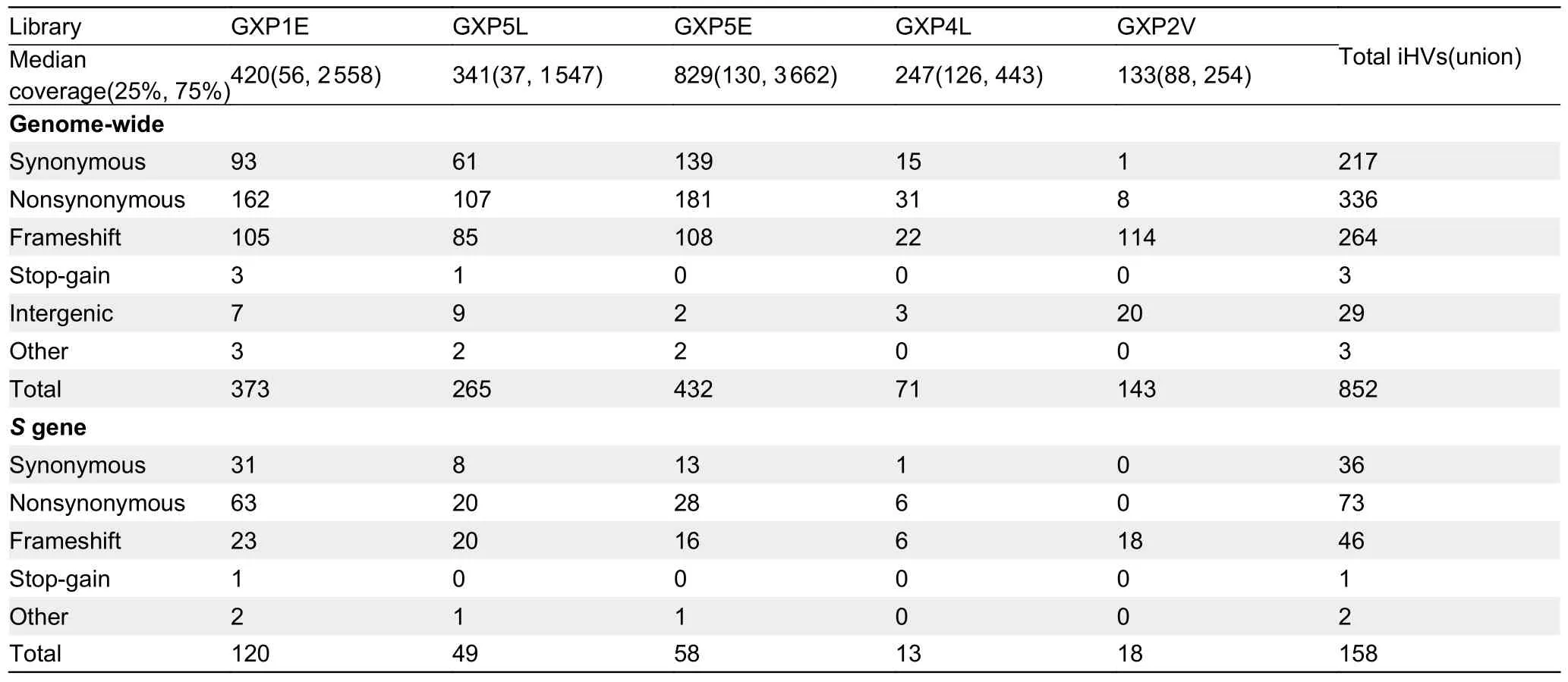
Table 1 The number of putative iHVs identified in the pangolin-CoV-GXC sublineage
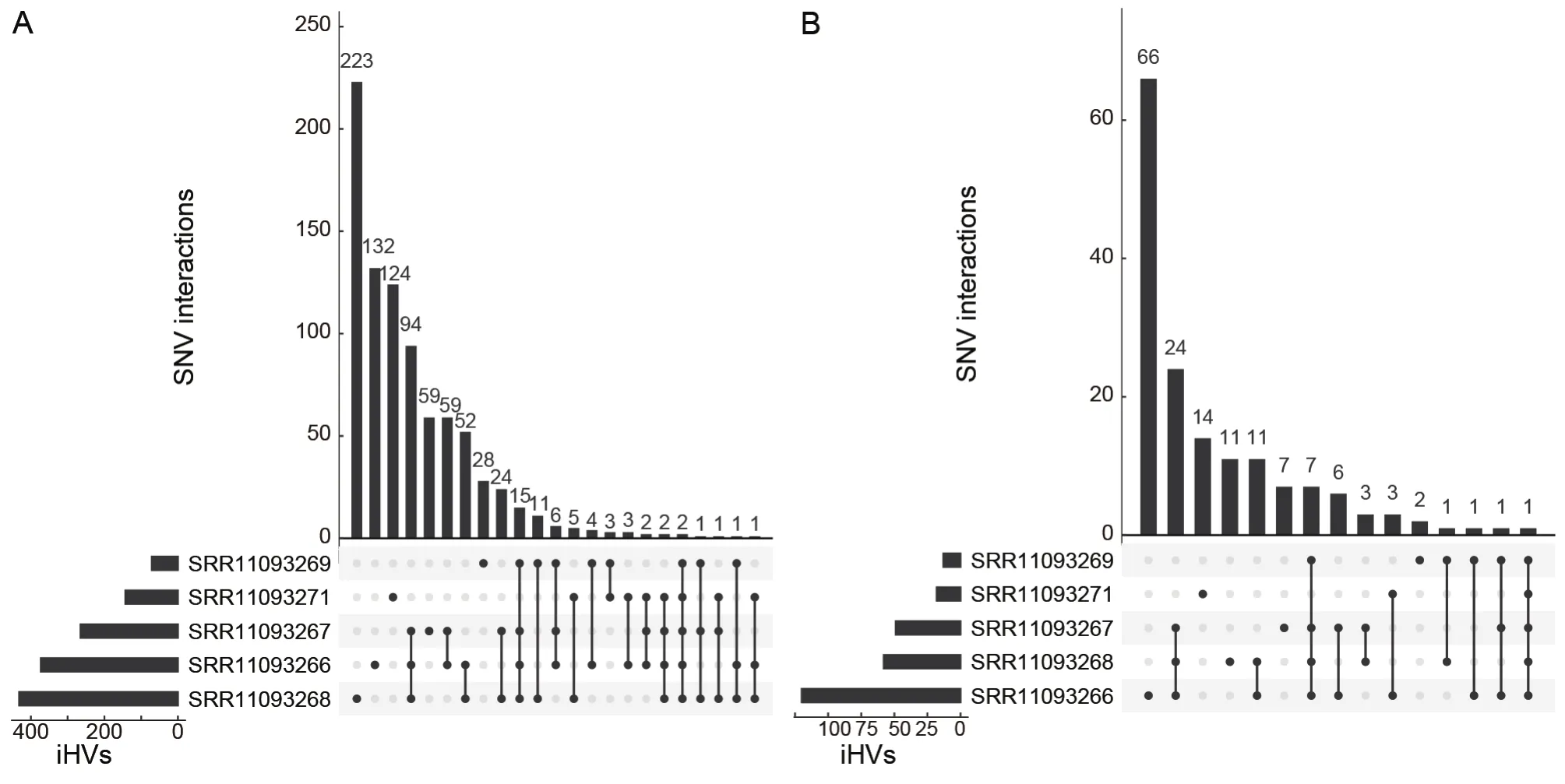
Figure 2 The numbers of overlapping iHVs between pangolin viral sequencing libraries at the genome-wide level (A) and at the level of the S gene (B)
Recombination events during Pangolin-CoV evolution
Since recombination is commonly observed in coronaviruses and is important for their evolution (Li et al., 2021; Sun et al.,2020; Wu et al., 2020), we asked whether different pangolin-CoV sublineages or strains have undergone recombination.We followed a previously reported strategy (Paraskevis et al.,2020) to screen putative recombination signals in the pangolin-CoV genomes and other SC2r-CoV and SARSr-CoV genomes.A putative recombination event involving a 346-nucleotide fragment (position 13666–14011 on the MP20 genome) in theORF1abgene was detected with the recombination detection program RDP4 (Martin et al., 2015)(Figure 3).The 346-nucleotide fragment, shared between MP20 and GXP5L, was likely introduced into the common ancestor of the pangolin-CoV-GXC sublineage from batderived SARSr-CoVs.Given the potential contribution of the spike protein RBD from the pangolin-CoV-GDC sublineage to SARS-CoV-2 via recombination (Liu et al., 2020; Xiao et al.,2020; Zhang et al., 2020), these results reveal that the occurrence of different recombination events in different pangolin-CoV sublineages further contributes to the diversity of pangolin-CoVs.
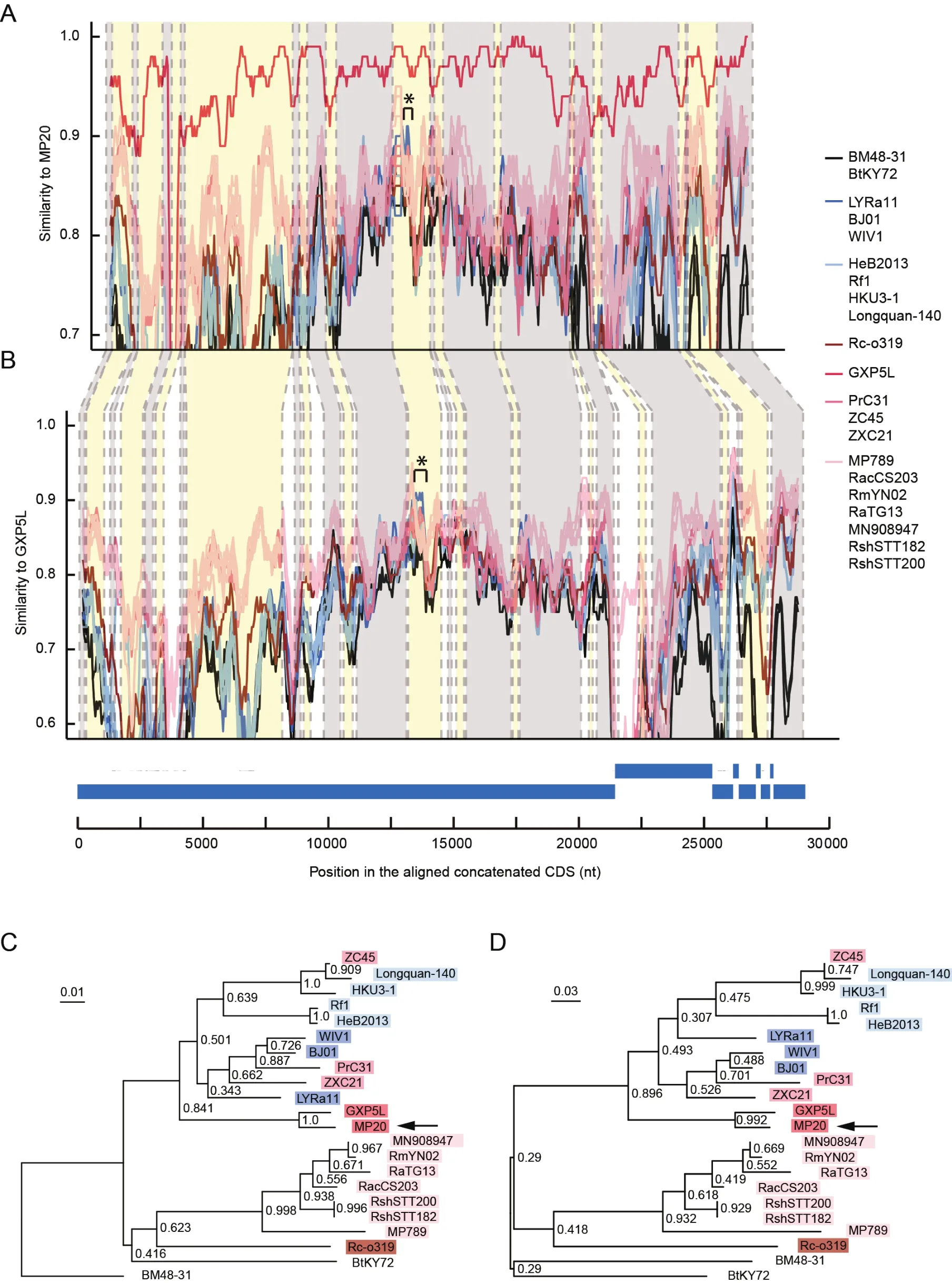
Figure 3 Recombination analysis of the pangolin-CoV-GXC sublineage
DISCUSSION
Due to the COVID-19 pandemic, coronaviruses as a research topic have garnered a tremendous amount of recent interest,and the literature contains a number of studies in which the roles of different animals in coronavirus transmission to humans are examined.Numerous studies have demonstrated that bats are rich reservoirs of the SC2r-CoVs (Hul et al.,2021; Murakami et al., 2020; Wacharapluesadee et al., 2021;Zhou et al., 2020a, 2020b, 2021).Nevertheless, a significant gap remains for the possible role of pangolins as a source of potential zoonosis of SC2r-CoVs, presumably due to the limited numbers of pangolins used to screen the SC2r-CoVs in previous studies or due to long postmortem intervals when the pangolins were confiscated in the illegal animal trades which typically lead to RNA degradation.Here, we applied the insolution hybridization capture sequencing to retrieve the genomic sequence of an SC2r-CoV from a highly degraded biomaterial.Technologies developed for the extraction and enrichment of nucleotides from ancient samples have the potential to build more complete viral genomes (Spyrou et al.,2019).Applying this strategy to massive collections in biobanks or museums will afford promising insights into the evolutionary history of SARS-CoV-2 and other SC2r-CoVs.
This study reveals the high level of genetic diversity of SC2r-CoVs hosted in pangolins and highlights the possibility that pangolins are an important natural host or reservoir of SC2r-CoVs.Both the pangolin-CoV-GDC and pangolin-CoVGXC viruses were isolated fromM.javanica(Malayan pangolin) in previous studies, and our results demonstrate for the first time that pangolin-CoV is also present inM.pentadactyla, suggesting that pangolin-CoVs have a relatively complex evolutionary history that deserves further study.It should be noted that both pangolin-CoV-GXC and pangolin-CoV-GDC had entry efficiencies into cells expressing the human ACE2 receptor comparable to those of SARS-CoV-2 pseudoviruses (Dicken et al., 2021; Guo et al., 2021; Nie et al., 2021; Niu et al., 2021; Zhang et al., 2021).Together, the observation of multiple recombination events during the evolution of SARS-CoV-2 (Boni et al., 2020; Gryseels et al.,2021; Zhou & Shi 2021), the high genetic diversity of pangolin-CoVs in Southeast Asia and neighboring regions, and the overlapping distributions of the pangolin-CoVs, bat-CoVs, and other coronaviruses (Lacroix et al., 2017; Latinne et al., 2020),highlight the ecological risk of zoonotic transmission of novel pathogenic SC2r-CoVs.The accumulated evidence demonstrates that pangolins should be included in the search for possible natural hosts or intermediate hosts of novel coronaviruses.
DATA AVAILABILITY
The raw sequencing reads generated in this study are available in the National Genomics Data Center (NGDC)Genome Sequence Archive (GSA) database (https://ngdc.cncb.ac.cn/gsa/) under BioProject accession No.PRJCA003816.The consensus sequence of pangolin-CoV MP20 has been deposited in the NGDC Genome Warehouse(GWH) (https://ngdc.cncb.ac.cn/gwh/) under accession No.GWHBEBQ00000000.The Sanger sequencing data were deposited in GenBank under accession Nos.MW173323,MW173324, and MW960369.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Y.P.Z., M.S.P.and J.L.conceived the project and designed the research.J.B.L.and R.N.W.conducted the wet lab experiments.J.N.Z.and J.J.T.provided an in-solution capture kit and protocol.H.L., Z.F.C., X.T., M.S.P., J.B.L., R.Y., J.Y.H.,Z.Y.Z., T.Z., and J.L.performed data analyses.T.T.Y.and R.N.W.prepared the samples.M.S.P., J.L., J.B.L., H.L.,Z.F.C., X.T., R.Y., and T.T.Y.drafted the manuscript with input from all authors.Y.P.Z., Z.G.Z., Z.L.S., Y.G.Y., L.Y., and X.M.L.revised the manuscript.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We thank Shi-Fang Wu and volunteers for the sampling work.We appreciate the suggestion for in-solution capture from Yun-Gui Yang, Ming-Kun Li, and Qiao-Mei Fu.We thank Wei Chen for comments on the manuscript.

- Zoological Research的其它文章
- Novel rhino-like SHJHhr mice with thyroid dysfunction
- A new species of Micryletta (Amphibia: Anura:Microhylidae) from the Langbian Plateau in southern Vietnam
- Site-specific and seasonal variation in habitat use of Eurasian otters (Lutra lutra) in western China:implications for conservation
- Understanding autism spectrum disorders with animal models: applications, insights, and perspectives
- High-quality chromosome-level genome assembly of redlip mullet (Planiliza haematocheila)
- Challenging Wallacean and Linnean shortfalls:Ectatosticta spiders (Araneae, Hypochilidae)from China