
Shifting equilibriums in Alzheimer's disease: the complex roles of microglia in neuroinflammation,neuronal survival and neurogenesis
2020-01-02 01:17SophieGrayKerriKinghornNathanielWoodling
中國(guó)神經(jīng)再生研究(英文版) 2020年7期
Sophie C. Gray, Kerri J. Kinghorn, Nathaniel S. Woodling
Institute of Healthy Ageing and Department of Genetics, Evolution and Environment, University College London, London, UK
Abstract Alzheimer's disease is the leading cause of dementia. Its increased prevalence in developed countries, due to the sharp rise in ageing populations, presents one of the costliest challenges to modern medicine. In order to find disease-modifying therapies to confront this challenge, a more complete understanding of the pathogenesis of Alzheimer's disease is necessary. Recent studies have revealed increasing evidence for the roles played by microglia, the resident innate immune system cells of the brain. Reflecting the well-established roles of microglia in reacting to pathogens and inflammatory stimuli, there is now a growing literature describing both protective and detrimental effects for individual cytokines and chemokines produced by microglia in Alzheimer's disease. A smaller but increasing number of studies have also addressed the divergent roles played by microglial neurotrophic and neurogenic factors, and how their perturbation may play a key role in the pathogenesis of Alzheimer's disease. Here we review recent findings on the roles played by microglia in neuroinflammation, neuronal survival and neurogenesis in Alzheimer's disease. In each case, landmark studies have provided evidence for the divergent ways in which microglia can either promote neuronal function and survival, or perturb neuronal function, leading to cell death. In many cases,the secreted molecules of microglia can lead to divergent effects depending on the magnitude and context of microglial activation. This suggests that microglial functions must be maintained in a fine equilibrium,in order to support healthy neuronal function, and that the cellular microenvironment in the Alzheimer's disease brain disrupts this fine balance, leading to neurodegeneration. Thus, an understanding of microglial homeostasis, both in health and across the trajectory of the disease state, will improve our understanding of the pathogenic mechanisms underlying Alzheimer's disease, and will hopefully lead to the development of microglial-based therapeutic strategies to restore equilibrium in the Alzheimer's disease brain.Key Words: adult neurogenesis; Alzheimer's disease; IGF-1; microglia; neuroinflammation; Trem2
Introduction
Alzheimer's disease (AD) is the predominant cause of late-onset dementia, and remains the last leading cause of death for which there is no disease-modifying therapy(Sperling et al., 2011). This lack of therapeutic progress can be attributed to AD's complex and heterogeneous aetiology,which has yet to be fully elucidated.
AD is characterized by a number of neuropathological hallmarks. The cardinal positive lesions include abnormal proteinaceous structures, namely β-amyloid (Aβ) plaques and neurofibrillary tangles, in addition to neuropil threads,dystrophic neurites, cerebral amyloid angiopathy and glial reactivity. The predominant negative lesions, neuronal death and synaptic loss, are strong correlates of cognitive impairment, and synaptic connections are thought to act as biological gateways to allow the propagation of neuropathology through the connectome of the brain (Seeley et al., 2009).The canonical wave of AD-specific neurodegeneration emanates through the entorhinal cortex and the Ammon's horn of the hippocampus, prior to encroaching upon the neocortex (Braak and Braak, 1995).
In an effort to understand the underlying risk factors and causes of AD, genome-wide association studies (GWAS) of AD have unearthed a number of loci at which allelic variants are associated with either an increased or a decreased risk of late-onset AD (Villegas-Llerena et al., 2016). The majority of the genes identified at risk loci are heavily implicated in immune system functions, such as the innate immune response, phagocytosis and lipid metabolism (Harold et al.,2009; Ferreira and Almeida, 2018). This has drawn attention to microglia, the tissue resident macrophages of the brain,which play a central role in neurodevelopment, central nervous system (CNS) homeostasis and ageing.
Microglia are the predominant modulators of neuroinflammation, a hallmark of AD pathology (Villegas-Llerena et al., 2016). From the initial days of classification, Aloysius Alzheimer observed that at AD presentation “the glia have developed numerous fibres and other morphological abnormalities” (Alzheimer et al., 1995). Since this observation,both genetic correlation studies in human populations andin vivomodelling have implicated microglia as cellular modulators of AD pathogenicity. Emerging clinical evidence also suggests a central role for microglia in AD. In the early AD brain, microglia are found in high densities surrounding Aβ plaques, and their presence in patients with mild cognitive impairment is associated with higher cortical grey matter and hippocampal volumes, in both Aβ-positive and Aβ-negative individuals (Femminella et al., 2019). Mechanisms proposed for this microglia-mediated neuropil preservation include the promotion of neurogenesis and neuronal survival, the endocytosis and degradation of extracellular debris and immunoresolution (Clayton et al., 2017). However, microglial reactivity also contributes to neurotoxicity,ranging from chronic neuroinflammation and aberrant synapse elimination to defective phagocytosis and reduced neurotrophic support (Mandrekar and Landreth, 2010; Tang and Le, 2016). These findings underscore the complexity of microglial responses in AD, where multiple factors cause an imbalance in microglial functions in neuroinflammation,neuronal survival and neurogenesis.
Search Strategy and Selection Criteria
For each topic addressed in this review, PubMed and/or Google Scholar were searched for papers with keywords matching that topic. Searches were carried out between February 2019 and October 2019. However, the authors acknowledge that this review is not a fully exhaustive analysis of all papers relevant to each topic, and apologise for any relevant work that could not be discussed due to space constraints.
Microglia and Neuroinflammation
In the AD microenvironment, the loss of microglial feedback systems can cause the balance of their pro- and anti-inflammatory actions to be shifted away from the physiological homeostatic equilibrium. Emerging evidence suggests that this chronic neuroinflammatory environment, and the activation of key innate immunity pathways, leads to neuronal compromise through the inhibitory effect on microglial Aβ clearance and the increase in neurotoxic factors that contribute to neuronal loss. However, there is equivocalin vivoevidence suggesting that some aspects of neuroinflammation, even under chronic conditions, continue to promote microglial phagocytosis and containment of Aβ, and thus have a neuroprotective function.
Microglial ontogeny and phenotypic diversity
In vivolineage tracing has revealed that microglia arise from erythromyeloid progenitors during embryological development and migrate from the yolk sac to the brain, where they proliferate and become nested within the CNS parenchyma(Ginhoux et al., 2010). During neurogenesis, microglia mediate circuitry rewiring through their influence on neuronal proliferation, differentiation and synaptic pruning (Tong and Vidyadaran, 2016). In healthy adult tissue, microglia are an extremely plastic and pleiotropic cell type. One helpful framework for describing the multiple states of microglial activation has been to categorise microglia in unchallenged(M0) and challenged (M1 or M2) states (Tang and Le, 2016).M0 microglia, also known as resting microglia, are active sentinel cells involved in the maintenance of a homeostatic CNS microenvironment (Nimmerjahn et al., 2005). Depending on the stimulus of activation, microglia can attain either an immunogenic phenotype, upregulating localized inflammation in response to disease or injury, or an anti-inflammatory phenotype. The latter promotes immunoresolution and tissue remodelling to minimize collateral damage inflicted by the immune response in the immune-privileged brain(Tang and Le, 2016). These polarized phenotypes have been definedin vitroas classically activated pro-inflammatory M1 microglia and alternatively activated M2 microglia, which promote immunoresolution and tissue repair (Tang and Le,2016) (Figure 1).In vivo, the phenotype of any individual activated microglial cell resides along the continuum between M1 and M2, which is reflected in the unique expression signature of cell-surface markers (Holtman et al., 2015).
M2 microglia exhibit a myriad of immunoprotective behaviours and promote CNS tissue repair and regeneration.They are subcategorized into three delineated activation states based on gene expression profiling (Satoh, 2018). M2a microglia arise via alternative activation and promote immunoresolution through the secretion of anti-inflammatory cytokines and phagocytosis of debris, and tissue regeneration through the production of neurotrophic factors such as insulin-like growth factor (IGF)-1. M2b microglia are activated by immune complex-mediated stimulation of Fc gamma receptors and are characterised by downregulated interleukin (IL)-12 secretion, in tandem with augmented IL-10 secretion (Holtman et al., 2015). M2c microglia emerge from the acquired deactivation of M1 microglia after exposure to glucocorticoids or IL-10. The M2c secretory profile is dominated by the expression of transforming growth factor beta (TGF-β) and sphingosine lipid kinase, which promotes tissue repair and matrix remodelling (Cherry et al., 2014).These distinctive cellular classifications were largely definedin vitroby their delineated differentiation protocols, and as with the previously polarized phenotypes,in vivomicroglial plasticity likely generates cells that less stringently adhere to categorized phenotypes, and are constantly adapting to the biological climate. Recent studies ofin vivomicroglia using single-cell RNA sequencing reveal that whilst the heterogeneity of myeloid cells in the brains of healthy mice decrease from embryonic stages of development to adulthood, in ageing and in demyelinating injury conditions, multiple distinct subpopulations of inflammatory microglia are found(Hammond et al., 2019; Li et al., 2019). Ongoing and future single-cell RNA sequencing studies from AD models, and from human brains, will likely provide a more accurate characterization of the multiple microglial states in the healthy and diseased brain.
Recent studies have begun to define the signaling molecules and growth factors supporting both resting and activated microglial functions. At rest, microglia are maintained by the activation of several key cytokine signaling axes, such as TGF-β and colony stimulating-factor 1 receptor, which can be secreted by surrounding neurons and astrocytes(Bohlen et al., 2017). These axes, as well as cell-autonomous activation programmes, are modulated in response to the detection of AD pathology to induce microglial reactivity and proliferation. In AD, activated microglia containing intracellular Aβ particles, in tandem with components of the complement cascade and immunoglobulins, have been discovered in proximity to senile plaques, and are enriched in the hippocampus of AD patients compared to cognitively normal controls (Bachstetter et al., 2015; Keren-shaul et al.,2017). Numerous ligands, receptors and signaling pathways influence microglial activation in these environments, some of which are discussed below.
Microglia and β-amyloid
AD is classified as a proteinopathy, due to its hallmark dysregulation of Aβ and tau proteostasis. The high density of microglia found around Aβ plaques is consistent with their role in Aβ clearance pathways and their activation by Aβ itself. Microglia possess a range of pattern recognition receptors (PRRs), including toll-like receptors, receptors for advanced glycation end products, and scavenger receptors,many of which can recognize different Aβ species through various interactions of differing affinity (EI Khoury et al.,1996; Landreth and Reed-Geaghan, 2009; Venegas and Heneka, 2017). Signaling downstream of PRR activation induces changes to cell surface marker expression, secretory profile and cytoskeletal organisation. Microglia can initiate receptor-mediated uptake and lysosomal degradation of oligomeric and protofibrillar Aβ, as well as expressing many highly active Aβ-degrading enzymes (including neprilysin,insulin-degrading enzyme, angiotensin-converting enzyme and matrix metalloprotease-9) (Zuroff et al., 2017). In the APP/PS1 mouse model of AD, microglia-mediated proteostasis significantly reduces neuropathology and cognitive impairment. However, these microglial activities are subject to an age-related, Aβ-contingent decline, coincident with a two- to five-fold downregulation in Aβ-degrading enzymes,namely neprilysin, insulin-degrading enzyme and matrix metalloprotease-9, as well as in scavenger receptors known to recognize Aβ, including receptors for advanced glycation end product, CD36 and SRA (Hickman et al., 2008; Zuroff et al., 2017). This downregulation of Aβ-binding receptors may be part of a larger, age-dependant transcriptomic shift in the microglial sensome, which is characterized by a downregulation in endogenous ligand recognition, in tandem with an overall upregulation of genes associated with neuroprotection (Hickman et al., 2013).
Microglia-associated genetic risk
The ontogenetically disparate lineage of microglia means they possess a transcriptomic profile that is distinct from other cells in the brain parenchyma. This has been exploited in the analysis of GWAS data, showing that a high proportion of AD-risk loci are selectively expressed in microglia in the brain (McQuade and Blurton-Jones, 2019). Whilst the biomechanisms behind many of these genetic risk factors are yet to be characterized, a few key protein-coding genes(includingAPOE,TREM2,CD33,PGRN,CR1andNLRP3)have been associated with disease-conferring pathways(Lambert et al., 2013; Dos Santos et al., 2017).
Microglia and apolipoprotein E
The ε4 allele of the apolipoprotein E (ApoE) gene was one of the first established genetic risk factors for AD, and it remains the most statistically significant (Liu et al., 2013).The ε4 allele is three-fold enriched in AD-patients compared to the general population, and homozygosity for ε4 confers an increased risk of AD from 20% to 90%, and a decreased mean age of onset from 84 to 68 years, compared to non-carriers (Corder et al., 1993).
ApoE is synthesized by astrocytes and microglia, and lipidated by ATP-binding cassette A1 to form lipoprotein particles (Holtzman et al., 2012). ApoE-lipoprotein acts as a chaperone for pathological species as it binds to both Aβ,in senile plaques, and to neurofibrillary ghost tangles, to enhance their rate of degradation through microglial receptor-mediated endocytosis and enzymatic activity (Jiang et al.,2008). The affinity of this binding is isoform-dependant, with the highest stability interactions completed by the protective ε2 allele and the ApoE2 isoform it encodes. Conversely, the AD-associated ε4 allele encodes the ApoE4 isoform, which has less stable, lower affinity binding to Aβ, and decreases Aβ internalisation by microglia (Tokuda et al., 2000). In accordance with this, in an AD patient cohort, the deposition of Aβ has been found to correlate positively with the number of ε4 alleles at the ApoE locus (Mann et al., 1997).
Microglia and triggering receptor expressed on myeloid cells 2
In addition to the risk conferred by ApoE ε4 alleles, GWAS studies have identified another set of risk alleles in theTREM2gene, encoding triggering receptor expressed on myeloid cells 2 (Trem2) (Guerreiro et al., 2013; Jonsson et al., 2013). Trem2 plays a highly pleiotropic role in microglial metabolic homeostasis, although its role in AD pathogenesis is yet to be fully elucidated (Zheng et al., 2018).
In AD mouse models, Trem2 has been shown to enhance microglia-plaque interaction through mediating microglial chemoattraction, and binding, to Aβ oligomeric species at nanomolar concentrations (Zhong et al., 2018). Trem2 is able to bind to all ApoE isoforms and enables microglia to engulf Aβ-lipoprotein complexes more efficiently than naked Aβ (Atagi et al., 2015; Bailey et al., 2015). Upon the binding of a ligand to Trem2, its intracellular signaling adaptor DAP12 is activated and phosphorylates downstream SYK and GSK3β, which in turn, can activate a multiplicity of signaling cascades (Zhao et al., 2018). In addition, DAP12 promotes the activation of chemotaxis, phagocytosis and the upregulation of key inflammasome components including NLR family pyrin domain containing 3 (NLRP3) (Heneka et al., 2013; Wang et al., 2015). This signalling axis also regulates microglial survival through the protein kinase B (PKB)/GSK3β-pathway-dependant increase in anti-apoptotic molecules such as Bcl-2, and microglial proliferation through the activation of canonical Wnt/β-catenin signaling (Zheng et al., 2017).
Homozygous loss-of-function mutations inTREM2have previously been associated with Nasu-Hakola disease, a rare autosomal recessive form of early onset dementia. Multiple independent studies have identified several keyTREM2lossof-function risk variants as being significantly associated with AD, the most common of which is the R47H variant,which increases the risk of AD threefold (Guerreiro et al.,2013; Yeh et al., 2017). The AD-associated Trem2 variants(R47H, R62H and D87N) all display impaired ligand-binding activity, in particular to Aβ oligomers and damage-associated apolipoproteins, and have a faster turnover rate,meaning they are less prevalent on the plasma membrane(Park et al., 2017). AD carriers of these loss-of-function variants present with more diffuse plaques and a far lower density of microglia clustering (Figure 2). The resulting insufficiency of microglial boundary formation is believed to allow increased diffusion of Aβ oligomers, which are highly toxic to surrounding neuronal populations (Yuan et al.,2016). Thus, Trem2 provides some of the strongest evidence for the protective capabilities of microglia in AD (Ulrich et al., 2017).
Microglia and CD33
Siglec-3 (CD33) is another microglial transmembrane protein known to regulate innate immunity (Crocker, 2002).One study showed that there was a significant increase in the levels of CD33-immunoreactive microglia in the frontal cortex of AD patients compared to age-matched controls,and that microglial expression of CD33 correlates with Aβ pathology (Griciuc et al., 2013). The minor rs3865444A allele ofCD33is neuroprotective and reduces the risk of AD development (Li et al., 2015). This AD-protective allele has two co-inherited single nucleotide polymorphisms (SNPs).The SNP in the promotor ofCD33acts as a proxy polymorphism for a secondary SNP that directly affectsCD33exon 2 splicing.In vivothese SNPs downregulate the expression of CD33, which was shown to significantly decrease the level of Aβ42in the AD brain (Griciuc et al., 2013). In culture,CD33-positive microglia have defective Aβ42uptake and degradative functions compared to CD33-negative microglia(Griciuc et al., 2013). Therefore, one neuroprotective mechanism of rs3865444A allele is likely to be enhanced microglia-mediated Aβ proteostasis.
In the 5xFAD transgenic mouse model of AD, knockout of CD33 reduces Aβ pathology and cognitive deficits in a TREM2-dependent manner, providing additional evidence for a detrimental role for CD33 in microglia (Griciuc et al.,2019). Transcriptomic profiling of microglia in these CD33 knockout mice revealed upregulated expression of genes involved in phagocytosis and cellular signaling, again in a TREM2-dependent manner (Griciuc et al., 2019). This interaction between CD33 and TREM2 underscores the fact that genetic risk factors do not act in isolation; instead they converge at the level of crosstalk in biological pathways. The elucidation of key axes involving multiple significant genetic risk factors for AD will therefore be essential for facilitating therapeutic development.
Microglia and other genetic risk loci
Other coding genes possessing risk-conferring polymorphisms, and encoding proteins critical to microglial function, include progranulin (involved in phagocytosis) (Minami et al., 2014) andNLRP3(Heneka et al., 2013; Tan et al.,2013). However, the majority of loci conferring significant risk are located in non-coding DNA, rendering their biomechanisms difficult to elucidate. A recent genome-wide assessment of the correlation between AD-risk variants and location of euchromatin structure in different cell types has revealed an enrichment of genetic risk loci in open chromatin in monocytic immune cells, namely macrophages and microglia (Tansey et al., 2018). These open chromatin sites contain consensus DNA-binding motifs for macrophage/microglial specific transcription factors, including SPI1 and MEF2 (Tansey et al., 2018). These results highlight a pivotal role for specific microglial transcriptional networks in polygenic AD risk.
Microglia and immunoresolution
Microglia are key regulators of the CNS microenvironment.Their highly dynamic and responsive secretory profile allows them to respond rapidly to extracellular signals to prevent damage to the immune-privileged brain. Acute inflammation can be immunoprotective through the neutralisation of pathogens or pathogenic peptides, such as Aβ, which reduces injury to the surrounding neurons. Under physiological conditions, following an acute inflammatory response, microglial signaling creates a negative feedback loop for effective resolution of inflammation. In models of brain injury,microglia initially upregulate their expression of IL-6 and other inflammatory cytokines, then produce elevated levels of IL-10 and TGF-β, with levels peaking 7 days post-insult to brain tissue (Taylor et al., 2017). Both IL-10 and TGF-β have a cardinal role in signaling to the surrounding microglia to mediate a regionalized shift to an immunosuppressive phenotype, re-establishing a homeostatic microenvironment(Qian et al., 2008; Kwilasz et al., 2015). TGF-β also promotes the production of neurotrophic factors and stimulates proliferative astrogliosis to reduce oligomeric Aβ-mediated synaptotoxicity (Diniz et al., 2017) (Figure 3). In addition to the role of microglia in AD, astrocyte proliferation is largely directed in a manner that preserves astroglial morphology,allowing the formation of a glial scar that minimises collateral damage inflicted by synaptotoxic Aβ species, promoting neural regeneration and network remodelling (Verkhratsky et al., 2013). In this way, microglial and astrocytic TGF-β signaling axes have been shown to reduce AD-associated dendritic spine loss in hippocampal neurons, and to increase performance in memory tasksin vivo(Diniz et al., 2017).
Disease-associated microglia
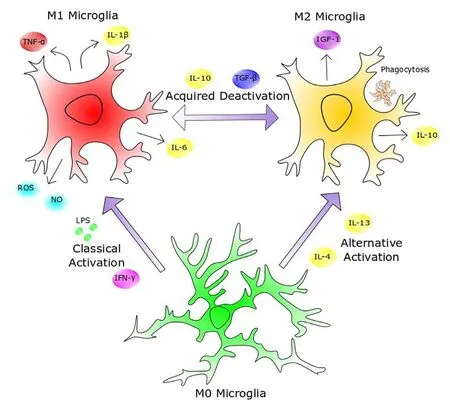
Figure 1 Heterogeneity of microglial activation.Sentinel (M0) microglia are highly active surveyors of their microenvironment, and upon detecting pathogenicity, such as through exposure to lipopolysaccharide (LPS) or interferon-γ (IFN-γ),they attain an M1 phenotype by inducing a highly inflammatory transcription profile and secreting pro-inflammatory cytokines, cytotoxic reactive oxygen species (ROS) and nitric oxide (NO).Once the disease or injury has been deemed resolved, the presence of immunosuppressive interleukin-10 (IL-10) and transforming growth factor-β (TGF-β) causes them to shift to the acquired deactivation M2 phenotype. The M2 phenotype can also be induced through alternative activation in the presence of IL-4 and IL-13, such as in the type 2 T helper cell secretory response.M2 microglia can be characterized by the secretion of anti-inflammatory cytokines and growth factors such as insulin-like growth factor 1 (IGF-1), in addition to their phagocytosis of extracellular debris. This plasticity of microglial phenotype forges a negative feedback loop wherein microglia can restrain the neuroinflammatory response within a critical window, below the threshold of self-toxicity, to allow rapid eradication of pathogenicity, whilst minimizing collateral damage to central nervous system parenchyma. TNF-α:Tumor necrosis factor-α.
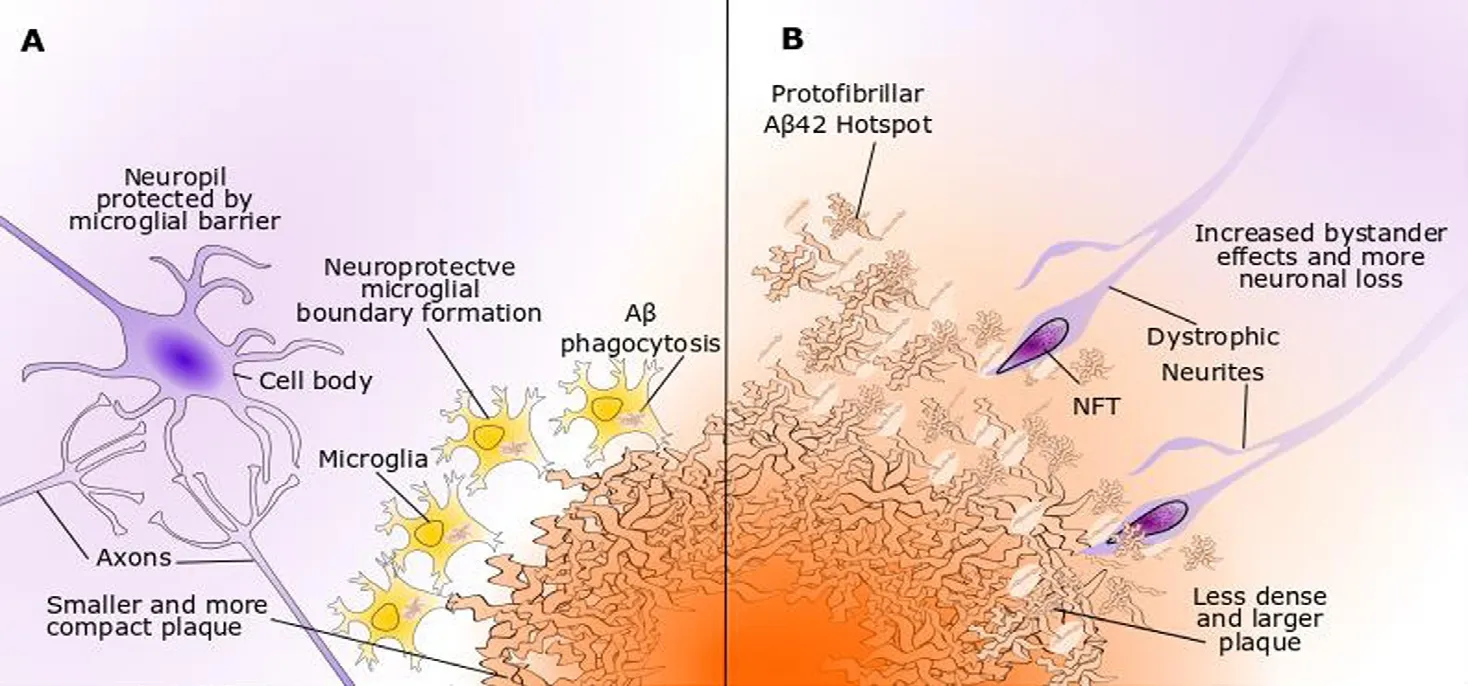
Figure 2 Trem2 and microglial responses to Aβ.Trem2 has nanomolar affinity for oligomeric Aβ species and enhances microglial chemotaxis to plaques in mouse models. (A)Microglia with wild-type Trem2 contribute to proteostasis through micropinocytosis of soluble Aβ oligomers and phagocytosis of Aβ fibrils. Microglial clustering around plaques forms a dense boundary to prevent outward diffusion of toxic Aβ and plaque expansion.(B) Microglia with a loss-of-function Trem2 variant display less efficient Aβ-directed chemotaxis, phagocytosis and boundary formation programmes. The plaques are less compact and show high seeding activity, leading to enlarged Aβ protofibrillar hotspots, higher inclusion of dystrophic neurites and increased neuronal loss. Aβ: β-Amyloid; NFT:neurofibrillary tangle.
Recently, single-cell transcriptomic analysis of murine models of AD has permitted the characterisation of a novel microglial phenotype termed disease-associated microglia(DAM) (Keren-shaul et al., 2017). It is suggested that chronic activation causes DAM to attain their phenotype through mechanistically coupled Trem2-independent and -dependent steps (Keren-shaul et al., 2017). The Trem2-independent step is characterized by the decreased expression of microglial homeostatic genes, such asTREM119, and the loss of inhibitory-checkpoint receptors, including CX3CR1 and CD200R, that ordinarily facilitate immunomodulation by interacting with neuron-bound and soluble cognate ligands(Hoek et al., 2000; Barclay et al., 2002). Thus, the loss of checkpoint inhibition means DAM are no longer receptive to inhibitory signals and attain a supraphysiological activated state. Following this, the Trem2-dependent pathway upregulates many genes encoding known AD risk factors, includingTREM2,APOEandTYROBP, and causes microglia to adopt a proinflammatory transcriptomic profile (Keren-shaul et al., 2017). This idea has been expanded in a recent review by Gomes-Leal, presenting the “friendly fire hypothesis”, wherein damage-associated molecular pattern molecules released by dying neurons in AD are capable of signaling through PRRs such as TLRs to activate the microglial immune response. The latter, if chronically activated, can lead to progression to a DAM phenotype (Gomes-Leal, 2019). Innate immune response mechanisms of reactive microglia, such as those of DAM, can become highly generalized and therefore may contribute to neuronal death through a bystander neurocytotoxic effect.
While DAM possess enhanced phagocytic programmes for Aβ uptake, they also can produce directly neurotoxic substances, including proinflammatory cytokines, such as IL-1β,IL-6 and tumor necrosis factor-α (TNF-α), as well as reactive oxygen species (ROS) through NADPH oxidase activation.At supraphysiological levels, NADPH oxidase production of ROS can cause oxidative stress and neuronal death. Even at levels that do not cause frank neuronal death, NADPH activation, when coupled with microglial complement receptor 3 activation and hypoxia, can induce long-term synaptic depression in hippocampal neurons (Zhang et al., 2014).
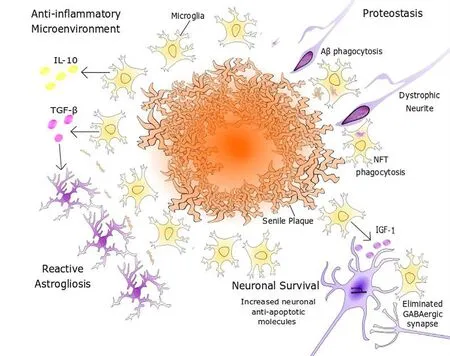
Figure 3 Promotion of neuronal survuval by microglia.Microglial expression of anti-inflammatory factors, such as IL-10, creates a milieu that is inductive of tissue repair and remodelling programmes. Microglial secretion of TGF-β can also induce astrocytes to undergo proliferative reactive gliosis to form a neuroprotective glial scar.Microglia promote neuronal survival through the secretion of neurotrophic factors, including IGF-1, and the elimination of axosomatic inhibitory synapses, thus upregulating neuron-endogenous synthesis of anti-apoptotic molecules such as Bcl-2 or Fgf2. Aβ: β-Amyloid; DCX: doublecortin; GFAP: glial fibrillary acidic protein;GN: granule neuron; IGF-1: insulin like growth factor-1; IL-10: interleukin 10; INP: intermediate neuronal progenitor; NFT: neurofibrillary tangle;QNP: quiescent neural progenitor; Sox2: sex determining region Y-box 2; TGF-β: transforming growth factor beta.
NADPH oxidase-induced ROS and mitochondrial ROS also activate NLRP3 and caspase-1, which are essential for the processing of pro-IL-1β into the mature and functional IL-1β form (Parajuli et al., 2013).In vivo, IL-1β can increase tau hyperphosphorylation and the formation of neurofibrillary tangles, through its activation of MAPK-p38, and can contribute to Aβ plaque formation (Griffin and Mrak, 2002;Sheng et al., 2001). However, IL-1β may also play protective roles in AD. For example, in the APP/PS1 mouse model of AD, overexpression of IL-1β in the hippocampus leads to reduced Aβ pathology and plaque formation (Shaftel et al.,2007). This shows that the influence of cytokine modulation upon microglial phenotype is extremely nuanced, with the fluctuation in one element of the cytokine-signaling network able to create seemingly antithetical downstream affects,contingent upon the experimental methodology used.
TNF-α can be secreted by both microglia and neurons and is a core component of the neuroinflammatory cascade,which like the preceding pro-inflammatory factors, can increase Aβ deposition and tauopathy over short periodsin vivo(Cavanagh and Wong, 2018). In keeping with this, the administration of TNF-α inhibitors in the TgCRND8 transgenic mouse model of AD, prior to Aβ plaque formation,prevented the manifestation of Aβ pathology and the associated increase in excitatory synaptic neurotransmission over a 4 month period (Cavanagh and Wong, 2018). However, the ablation of TNF-α receptor expression in triple-transgenic AD model mice depleted microglial phagocytic markers and endocytic activity, leading to elevated Aβ deposition and tau-related pathology (Montgomery et al., 2011). These findings suggest that the net effect of TNF-α signaling undergoes inversion during the trajectory of AD. In the preclinical stages of AD, proinflammatory TNF-α signaling may contribute to pathological progression, whilst in the clinical stages,TNF-α may become neuroprotective, due to its critical role in microglial-mediated Aβ clearance.
Taken together, these studies illustrate the complexities that arise when attempting to decode the role of DAM in AD pathogenesis, where even a single signaling axis can produce seemingly disparate experimental results. Perhaps equally importantly, these studies highlight that most microglial functions exist in a state of equilibrium in the healthy brain, and that interventions or disease processes, such as those present in AD, that disrupt this equilibrium in either direction, can produce detrimental effects on neuronal health and survival.
Microglia and Neuronal Survival
Microglia are key to maintaining the neuron population and regulating neuronal survival. As shown above, microglia can indirectly modulate neuronal survival through their proteostatic and immunomodulatory mechanisms (Figure 3).However, studies bothin vivoandin vitrohave shown that microglial secreted factors can directly regulate neuronal survival. Moreover, microglia-mediated circuitry rewiring,in the form of eliminating inhibitory synapses, has been found in murine models of innate immune activation, and is thought to influence neuronal survival (Chen et al., 2014).
Microglial insulin-like growth factor and neuronal survival
The effect of IGF signaling on neural cell growth and survival is highly complex and under physiological conditions can be seen as an evolutionarily conserved system for resource allocation (Fernandez and Torres-alemán, 2012). IGF-1 signaling is thought to promote microglial mitogenesis and a shift to an M2 phenotype, and has a complex and controversial involvement in neuroinflammation (Labandeira-garcia et al., 2017).
In vivo, microglial secretion of IGF-1 can directly pro-mote hippocampal CA1 neuronal survival through IGF-1 receptor-mediated activation of the phosphatidylinositol 3-kinase (PI3K)-PKB signalling axis (Wine et al., 2009). In AD patients, neurons of the hippocampus, and to a lesser extent those of the cerebellar cortex, show a significant reduction in the responsiveness of their IGF-1R/IRS-2/PI3K signaling pathway, which is associated with cognitive decline and disease progression (Talbot et al., 2012). In this way,the inability of neurons to effectively transduce microglial neurotrophic signaling factors could play a major role in the reduced resistance to the proteostatic dysregulation and cytotoxic inflammatory signals of AD (Kandimalla et al.,2017). Consistent with this idea, therapeutic upregulation of the downstream PI3K-PKB signaling pathway through CNS intranasal insulin treatment has been found to enhance memory and cognition in both murine models and in limited human trials (Craft et al., 2017; Lochhead et al., 2019).
However, IGF-1 signaling has also been implicated in ageing and proteostatic failure, with reduced IGF-1 and/or insulin signaling being a phylogenetically conserved mechanism for increasing healthy lifespan in animal models from worms to mice (Steculorum et al., 2014; Augustin et al., 2017).Consistent with this proposed detrimental role for IGF-1 signaling in AD, heterozygous deletion of the IGF-1 receptor partially rescues spatial memory impairment and synaptic loss in AD mouse models, potentially due to an associated reduction in soluble Aβ oligomers (Cohen et al., 2009).
Reduced neuronal IGF-1 signaling may have additional beneficial roles in relation to endosomal recycling pathways that decline with age. In cultured human cells, chronic moderate reduction in IGF-1 signaling stimulates endosomal recycling of gap junction proteins by increasing the activity of specific small GTPases (Augustin et al., 2017). In theDrosophilagiant fibre system, chronically reduced insulin-like signaling conserves gap junction density with age,and rescues the age-associated decline in neurotransmission in a proteasome-dependent manner (Augustin et al., 2017,2018). Consistent with this finding, chronic reduction of insulin-like signaling inDrosophilaneurons is sufficient to extend healthy lifespan (Augustin et al., 2018).
Thus, pathological perturbation to a pathway as multifaceted as the IGF-1 signaling axis can have downstream effects that are differentially neuroprotective and neurotoxic.Moreover, it suggests that whilst life-long chronic inhibition of the IGF-1 pathway is associated with protection against neuronal ageing, in individuals with AD, an acute increase in microglial IGF-mediated neurotrophic support, or the responsiveness of neurons to IGF stimulation, could potentially protect against neuronal death.
Microglia-mediated elimination of GABAergic synapses
At glutamatergic synapses, N-methyl-D-aspartate type glutamate receptors (NMDARs) are enriched on pyramidal neurons in both the hippocampus and the cortex, and play an important role in glutamate-mediated synaptic plasticity and neuronal survival. In organotypic hippocampal slices, Aβ oligomers act through an NMDAR-dependant mechanism to decrease dendritic spine density and trigger progressive loss of active glutamatergic synapses in pyramidal neurons(Shankar et al., 2007). The consequent reduction in synaptic plasticity, neuronal death and synaptic loss has been suggested to be the basis for the onset and progression of cognitive impairment and memory deficits in AD (Knobloch and Mansuy, 2008).
The use of three-dimensional electron microscopy and electrophysiology has shown that in adult mice, following injury, microglia can indirectly upregulate survival pathways in cortical neurons through eliminating inhibitory synapses(Figure 3). Microglia extend their processes into the cleft between the pre-synaptic and post-synaptic components of axosomatic synapses to detach their connection and prevent GABAergic signalling (Chen et al., 2014). This has a disinhibitory effect on cortical neurons, which increases their firing rate and leads to NMDAR-mediated plasticity in the postsynaptic excitatory synapses (Chen et al., 2014).NMDARs mediate increased Ca2+influx into the soma cytosol, activating CaM kinase IV, which phosphorylates CREB,allowing it to promote the transcription of multiple target genes encoding anti-apoptotic and neurotrophic molecules(Sakamoto et al., 2013). Elevated NMDAR signaling activity can also upregulate the expression of the NFI-A transcription factor, which activates downstream neuronal nitric oxide synthase-and mitogen activated protein kinase-dependant survival pathways (Zheng et al., 2010).
However, if this NMDAR-mediated Ca2+influx becomes excessive, it can promote glutamatergic excitotoxicity, which is implicated in the preferential loss of neocortical, cortical and hippocampal pyramidal neurons in AD. Supraphysiological Ca2+concentrations can instigate somatodendritic compartment swelling, mitochondrial dysfunction, free radical generation, tau phosphorylation and synaptic loss (Liu et al., 2019). This deleterious excitotoxic cascade leads to the instigation of apoptotic and necroptotic programmes in cortical and subcortical neurons (Caccamo et al., 2017).
Microglia can thus promote neuronal survival through the production of growth factors such as IGF, or through synaptic remodelling, to promote NMDAR-mediated survival pathways. However, chronic overproduction of growth factors, or excessive synaptic remodelling, can also have detrimental consequences. As is true for their complex roles in modulating neuroinflammation, the effects of microglia on neuronal survival exist in an equilibrium, with building evidence pointing to the disruption of that equilibrium as a key event in AD pathogenesis.
Microglia and Adult Hippocampal Neurogenesis
In addition to providing support for the survival of mature neurons, microglia have a central role to play in modulating the production of new neurons in the adult brain. Neural stem cells (NSCs) depend on signals from their niche, including microglia, to regulate their self-renewal, proliferation and differentiation. In the absence of microglia, NSCs progressively lose the capacity to undergo the differentiation process required for neurogenesis (Walton et al., 2006). The role of microglia in neurogenesis can be seen as instructive,with microglial-secreted factors such as IGF-1 and trypsinogen having the capacity to regulate adult NSC proliferation and differentiation (Nikolakopoulou et al., 2013; Mir et al.,2017). Thus, the shift in microglial phenotype that occurs over the AD trajectory is likely to have an important and extremely complex impact on adult neurogenesis.
Cascade of adult hippocampal neurogenesis
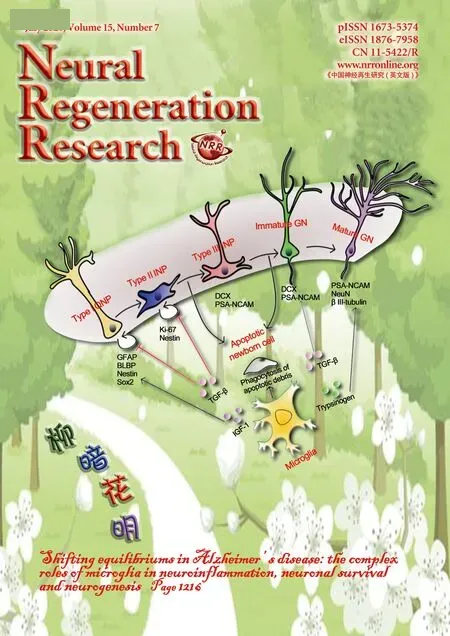
Research with rodent models has revealed two distinct brain regions, namely the subventricular zone and the hippocampal dentate gyrus (DG), as centres of adult neurogenesis,with DG neurogenesis most well-studied in AD. In the subgranular zone of the DG, type I quiescent neural progenitors (QNPs), characterised by their expression profile of glial fibrillary acid protein (GFAP), sex determining region Y-box 2 (Sox2) and brain lipid-binding protein, give rise to new neurons through a neurogenic cascade of asymmetric divisions and differentiation (Encinas et al., 2011; Gon?alves et al., 2016). The majority of new-born neural progenitors undergo apoptosis, and microglia phagocytose the apoptotic debris from these cells to help maintain the equilibrium of the neurogenic niche (Sierra et al., 2010). QNPs asymmetrically divide to allow self-renewal and the production of type II intermediate neuronal progenitors (INPs), expressing Ki-67 and Nestin. Type II INPs then differentiate into type III neuroblast INPs, which undergo further lineage restriction to generate immature and mature granule neurons (GNs)(Encinas et al., 2011; Gon?alves et al., 2016).
In mouse models, genetic risk factors associated with neurological disorders can bidirectionally disrupt normal adult hippocampal neurogenesis (AHN) in a microglial-dependant manner; either through the failure of microglial-dependant apoptosis of immature GNs, or the loss of microglial-mediated progenitor proliferation (Appel et al., 2018).
Human Hippocampal Neurogenesis in Alzheimer's Disease
Animal models of neurogenesis from rodents to non-human primates display AHN, which is subject to an age-dependant decrease that varies across phylogeny (Kuhn, 2015).However, the nature of AHN in humans remains to be fully elucidated and is subject to much scientific debate. An early study by Eriksson et al. (1998) demonstrated the presence of dividing progenitor cells and new neurons in the DG of post-mortem adult brain tissue, with subsequent reports arising both in support and in rebuttal of these findings. Recently, the publication of three reports in quick succession have presented contrasting evidence concerning the presence of human AHN.
In one recent report, Sorrells et al. (2018) analysed hippocampal DG samples from post-mortem brain tissue of individuals from 14 gestational weeks to 77 years old. They found a rapid and age-dependant depletion in hippocampal neurogenesis, which became undetectable beyond 13 years of age, a finding that the authors highlighted to be unique in humans.
In another recent study, Boldrini et al. (2018) quantified neuronal progenitor cells in autopsied hippocampi from healthy human individuals between 14 and 79 years of age.Their study showed that whilst the abundance of type I neural progenitor cells decreases with ageing, type II intermediate neuronal progenitors remain stable. Although both studies employed similar immunohistochemical methodology, there were significant differences in the age range and inclusion criteria of brain tissue samples. In addition, there was variation in stringency to post-mortem delay period,which can affect the immunoreactivity of biomarkers, such as doublecortin (DCX) used in both studies to identify immature neurons (Boekhoorn et al., 2006). This could explain the differences in outcomes of the two studies. Another recent study by Moreno-Jimenez and colleagues, using stringent sample exclusion criteria, and an innovative tissue processing approach, found thousands of DCX positive neurons in the DG of 13 neurologically healthy adult brain tissue samples, which were absent from proximal non-neurogenic regions (Moreno-Jiménez et al., 2019). However, in samples from AD brains, there was a decrease in both the number and the maturation of neurogenic precursors as the disease progressed. This highlights AHN as an important potential pathological correlate of AD, meriting further investigation,specifically in the context of changes to microglial reactivity in AD, which has been shown to be associated with disrupted neurogenesis (Appel et al., 2018).
Microglial activation and neurogenesis
As previously discussed, in AD, microglia adopt a spectrum of active phenotypes. In the study of some neurological disorders it has been suggested that overactive microglia might be inhibitory of AHN (Ekdahl et al., 2003; Pluchino et al.,2008; Tepav?evi? et al., 2011). For instance, microglial-mediated neuroinflammation can be disruptive to neurogenic niches, and undermine the integrity of neuronal population replenishment programmes (Fan and Pang, 2017). Both acute and chronic microglial activation can modulate neurogenesis.In vitro, microglia acutely activated with LPS strongly express IL-1, IL-6 and TNF-α, which reduces neural progenitor cell survival. Conversely, microglia chronically activated by LPS, with a secretory profile dominated by IL-10 and prostaglandin E2, are highly permissive to the neurogenic cascade (Cacci et al., 2008). This suggests that the complex set of molecules secreted from microglia in different activation states can both promote and inhibit neurogenesis depending on the context.
Some microglial neurotrophic signals, such as IGF-1 and trypsinogen, have been shown to promote neurogenesis (Nikolakopoulou et al., 2013; Mir et al., 2017). The secretion of trypsinogen by activated microglia was found to increase the generation of neuron-specific class III beta-tubulin positive cells (Nikolakopoulou et al., 2013). IGF-1 activates Ras-related GTPase activity in QNPs, which allows PKB-dependant phosphorylation and stabilisation of the neuron-determining Sox2 master regulatory transcription factor (Mir et al.,2017). Once stabilised, Sox2 becomes transcriptionally active and enhances the expression of its pro-neural gene targets to initiate downstream neurogenic differentiation programmes.However, during the trajectory of disease progression in transgenic murine models of AD, SGZ cell proliferation is depleted despite microglial secretion of IGF-1 remaining constant (Myhre et al., 2019).
Other microglia-derived factors such as TGF-β appear to have more complex effects on neurogenesis. In a murine ME7 model of prion disease-mediated neurodegeneration,microglial proliferation in the DG positively correlates with increased hippocampal neurogenesis, and a candidate gene screen revealed TGF-β as the molecular correlate of microglial-dependent neurogenesis (De Lucia et al., 2016). In addition, TGF-β has the capacity to promote neurogenesisin vitro, and blockade of TGF-β signaling reduces the percentage of PSA-NCAM positive cellsin vivo(Battista et al., 2006).However, TGF-β signaling can also reduce proliferation in Sox2/GFAP-expressing cell compartments of the SGZ and promotes their shift into the G0 phase (Wachs et al., 2006).Thus, although TGF-β may promote the later stages of neuronal differentiation and neuronal survival, chronic TGF-β signaling may reduce the pool of neural stem cell progenitors in the SGZ, potentially explaining the long-term reduction in neurogenesis that is posited to occur in AD.
Microglial factors may also explain some effects of familial AD genes on neurogenesis. A risk variant at Presenilin 1(PSEN1) is one of the three loci linked to familial AD. Neural progenitor cells from wild-type and PS1-risk variant expressing mice demonstrate no significant difference in proliferation and differentiation. However, wild-type neural progenitor cells co-cultured with PSEN1 risk-variant expressing microglia, or mutant-microglia conditioned medium, displayed deficits in proliferation and progression along the neurogenic cascade (Choi et al., 2008). This suggests that microglial secretions are a key regulator of AHN, and may imply that one effect of the PSEN1-risk variant in familial AD is the impairment of microglial-mediated support of AHN.
Finally, microglial phagocytosis of apoptotic debris can modulate adult neurogenesis. In the SGZ, the majority of new-born neural progenitor cells undergo apoptosis 1-4 days after they are generated (Sierra et al., 2010). Non-activated microglia play an important role in maintaining homeostasis in the SGZ neurogenic niche by phagocytosing apoptotic new-born cells. This microglial phagocytic activity is apparently unchanged by ageing or acute neuroinflammation, suggesting it is a mechanism that promotes a homeostatic neurogenic niche in both healthy and disease states(Sierra et al., 2010). However, the possibility remains that chronic activation of microglia in AD may exert different effects on neurogenesis.
Taken together, this experimental evidence implies that neither microglial activation, nor neuroinflammation, are definitively pro or anti-neurogenic in nature, and that it is the unique cytokine and neurotrophic profiles of microglia that determine their effect on neurogenesis and tissue repair(Figure 4). In the healthy brain, this profile maintains a permissive equilibrium for appropriate levels of neurogenesis,but changes in microglial inflammatory and neurotrophic signals have the capacity to disrupt neurogenesis in AD and other neurodegenerative diseases.
Conclusion
There exist immense complexities when attempting to unravel the roles of microglia in neuroinflammation, neuronal survival and neurogenesis in AD. Progress towards a more systematic understanding of microglia in AD is being made through experimental approaches that integrate data from multiple levels, and which appreciate the biological intricacies of neurodegenerative diseases. One area that merits additional study is the changing role of microglia across the trajectory of AD pathogenesis, as stage-dependent microglial phenotypes may be poorly modeled by manyin vitroand animal models of AD.
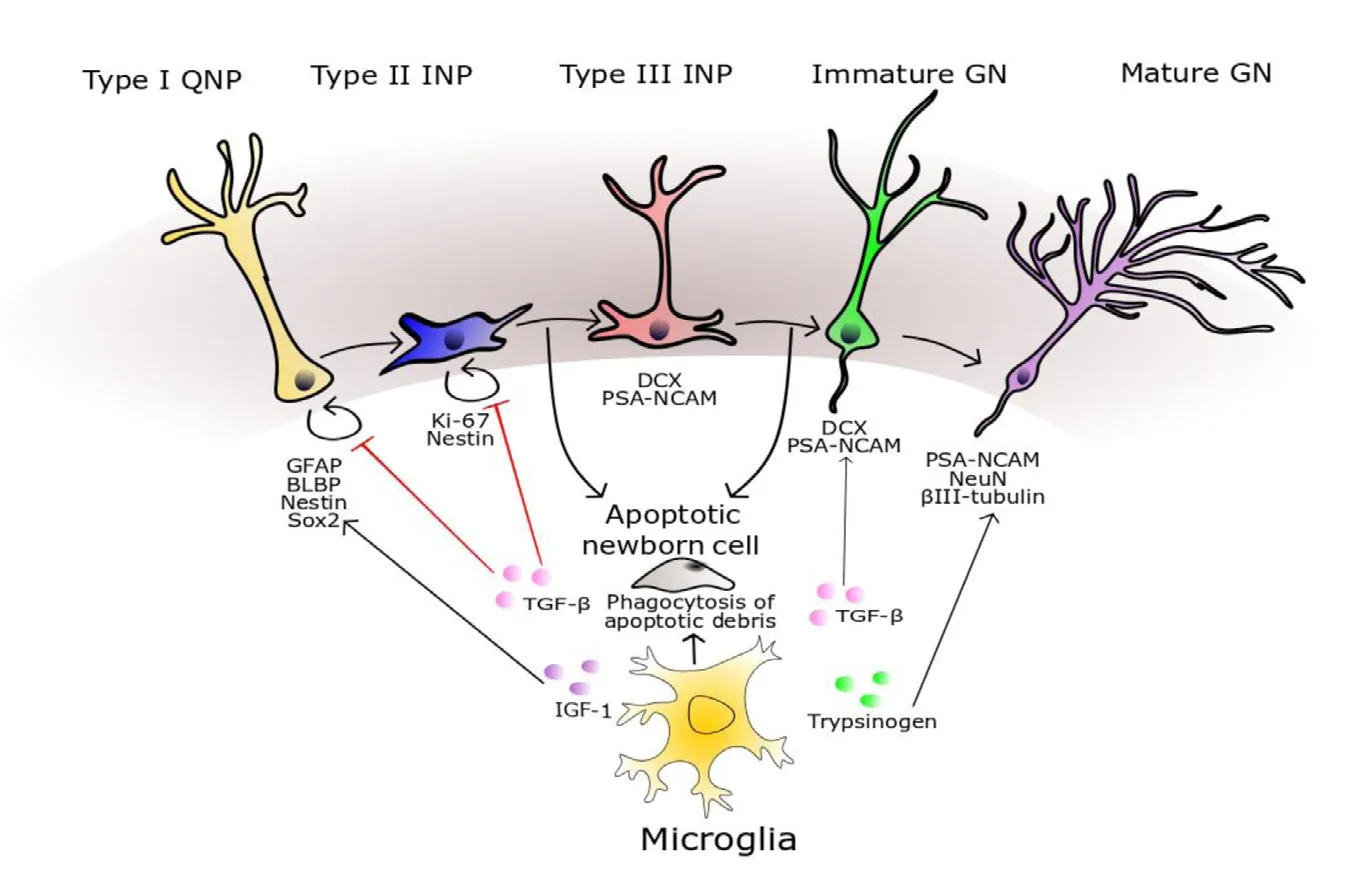
Figure 4 Modulation of adult neurogenesis by microglia.Microglial phagocytosis of cellular debris from apoptotic neural progenitors is an important homeostatic mechanism in the neurogenic niche. Type II INPs express Ki-67 and Nestin, whilst Type III INPs and immature GNs express DCX and polysialylated neural cell adhesion molecule (PSA-NCAM). Microglial secretion of TGF-β has a complex effect on neurogenesis, with one hallmark effect being to promote the differentiation of neural progenitors to PSANCAM positive cells. The transcriptomic profile of mature GNs is characterised by the expression of neuronal nuclear marker (NeuN), Prox-1, calbindin,and neuron-specific class III beta-tubulin (βIII-tubulin). Microglial secretion of trypsinogen augments the number of βIII-tubulin-positive cells, potentially through a mechanism functioning upstream in the neurogenic cascade. BLBP: Brain lipid binding protein; DCX: doublecortin; GFAP: glial fibrillary acid protein; GN: granule neurons; IGF-1: insulin like growth factor; INP: intermediate neuronal progenitor; QNP: quiescent neural progenitors; Sox2: sex determining region Y-box 2; TGF-β: transforming growth factor beta.
In many publications, terms such as ‘double-edged sword'are utilized to help understand paradoxical data on the role of a biological component of AD pathogenesis. However, a complementary framework may be to describe these components as existing in a biological state of equilibrium in the healthy brain. The displacement of these components beyond the physiological parameters of this equilibrium can therefore lead to pathological consequences, as are seen in AD. The development of effective microglia-based disease-modifying therapies will therefore be contingent upon understanding the heterogeneity of microglial phenotypes,as their disequilibrium impacts upon neuroinflammation,neuronal survival and neurogenesis both in the healthy brain and in AD.
Acknowledgments:The authors would like to thank the Wellcome Trust, the Academy of Medical Sciences, the Rosetrees Trust (KJK) and the Alzheimer's Society (NSW) for their kind support. SCG also gratefully acknowledges the support of Professor Leslie Dale, UCL.
Author contributions:Writing original draft: SCG; writing review and editing: SCG, KJK, and NSW; figures and visualisation: SCG. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Larry Baum, The University of Hong Kong, China.
Additional file:Open peer review report 1.
 中國(guó)神經(jīng)再生研究(英文版)2020年7期
中國(guó)神經(jīng)再生研究(英文版)2020年7期
- 中國(guó)神經(jīng)再生研究(英文版)的其它文章
- Glial cells in intracerebral transplantation for Parkinson's disease
- Fast-tracking regenerative medicine for traumatic brain injury
- The N-formyl peptide receptors: contemporary roles in neuronal function and dysfunction
- Adrenomedullin: an important participant in neurological diseases
- ABC efflux transporters at blood-central nervous system barriers and their implications for treating spinal cord disorders
- Biomaterials and neural regeneration