
gFACs: Gene Filtering, Analysis, and Conversion to Unify Genome Annotations Across Alignment and Gene Prediction Frameworks
2019-10-22 08:51:52MadisonCaballeroJillWegrzyn
Madison Caballero*, Jill Wegrzyn*
Department of Ecology and Evolutionary Biology, University of Connecticut, Storrs, CT 06269, USA
KEYWORDS Genome annotation;Bioinformatics;Protein annotation;Gene prediction;Alignment
Abstract Published genomes frequently contain erroneous gene models that represent issues associated with identification of open reading frames, start sites, splice sites, and related structural features.The source of these inconsistencies is often traced back to integration across text file formats designed to describe long read alignments and predicted gene structures.In addition,the majority of gene prediction frameworks do not provide robust downstream filtering to remove problematic gene annotations, nor do they represent these annotations in a format consistent with current file standards. These frameworks also lack consideration for functional attributes, such as the presence or absence of protein domains that can be used for gene model validation.To provide oversight to the increasing number of published genome annotations, we present a software package, the Gene Filtering,Analysis,and Conversion(gFACs),to filter,analyze,and convert predicted gene models and alignments. The software operates across a wide range of alignment, analysis, and gene prediction files with a flexible framework for defining gene models with reliable structural and functional attributes. gFACs supports common downstream applications, including genome browsers, and generates extensive details on the filtering process,including distributions that can be visualized to further assess the proposed gene space. gFACs is freely available and implemented in Perl with support from BioPerl libraries at https://gitlab.com/PlantGenomicsLab/gFACs.
Introduction
In the era of high-throughput sequencing, the size and complexity of the genomes assembled in recent years,have dramatically increased.Despite this,only a handful of the nearly 7800 eukaryote genomes in GenBank are resolved at, or close to,chromosome level[1].In addition,over 85%of these genomes contain some type of gene annotation errors [2-4]. These challenges are likely to persist with projects, such as the Earth BioGenome Project, planning to sequence 1.5 millions of eukaryotic genomes in coming years [5]. Initiatives such as these will assemble and annotate increasingly large and complex genomes to assess greater biodiversity.
The majority of genome annotations are semi-automated,derived from informatic approaches that involve a combination of sequence alignments and ab initio predictions[6-8].The inputs may include pre-assembled transcripts,raw RNA-seq reads,and closely related proteins.The resources considered depend on the available evidence,as well as the complexity and size of the genomes under investigation.The downstream genome annotations and upstream alignment files are represented in one of the more variable bioinformatic standard file formats,known as the Generic Feature Format(GFF)[9].The GFF file provides structure for information-rich annotations as compared to the reduced representation available through the General Transfer Format(GTF).Generation of a final gene annotation requires filtering of incomplete or unlikely structural models and consideration of functional annotations at the full protein or protein domain level.The informatic packages that distill several sources of evidence into gene annotations, frequently deliver these without tools to assess their validity.
The Gene Filtering,Analysis,and Conversion(gFACs)represents a flexible annotation refining application that can accept standard annotations from primary gene annotation software as well as transcript/protein sequence aligners. In combination with the reference genome,gFACs can filter erroneous gene models, generate statistics/distributions, and provide outputs for standard downstream processing and/or visualization. Notably, this application does not replace the ab initio or similarity-based prediction models, but serves as a companion tool to resolve conflicting annotations and improve the quality of the final models. gFACs is unique in its ability to provide statistics and analysis,along with a direct connection to functional annotations to refine models.Similar programs such as gffread and gffcompare (https://ccb.jhu.edu/software/stringtie/gffcompare.shtml) provide gene model filtering and comparison abilities but lack application for analysis such as comprehensive statistics, functional annotation inclusion, and output standardization. gFACs can be used in tandem with these tools as it recognizes gffread inputs and provides a compatible GTF output for both gffread and gffcompare. The goal of gFACs is to aid the users in understanding their data while providing customized filters and utilities to remove and analyze gene models.As novel genomes are annotated,flexible customization and tools for analysis are essential for the tuning of final models.
Method
Accepted inputs span a range of aligners and gene predictors,which are presented in formats with similarities to GTF and GFF files. Current accepted input formats include MAKER[6],Prokka[7],BRAKER/AUGUSTUS[8],EVidenceModeler[10], GMAP [11], GenomeThreader [12], gffread, Exonerate[13], and NCBI GFF annotations. The user specifies the file source at runtime,which can be selected from an applicable set of flags. gFACs can optionally accept the reference genome in FASTA format(standard or softmasked)to permit more refined filtering and analysis.The second optional file is the annotation flat file resulting from EnTAP[14],which provides a functional annotation summary, including similarity search, protein domain, and gene family assignments for the proposed gene models or aligned sequences (Figure 1). The physical positions represented in these files are formatted into an intermediate text file to aid in processing and calculating the proposed gene space.
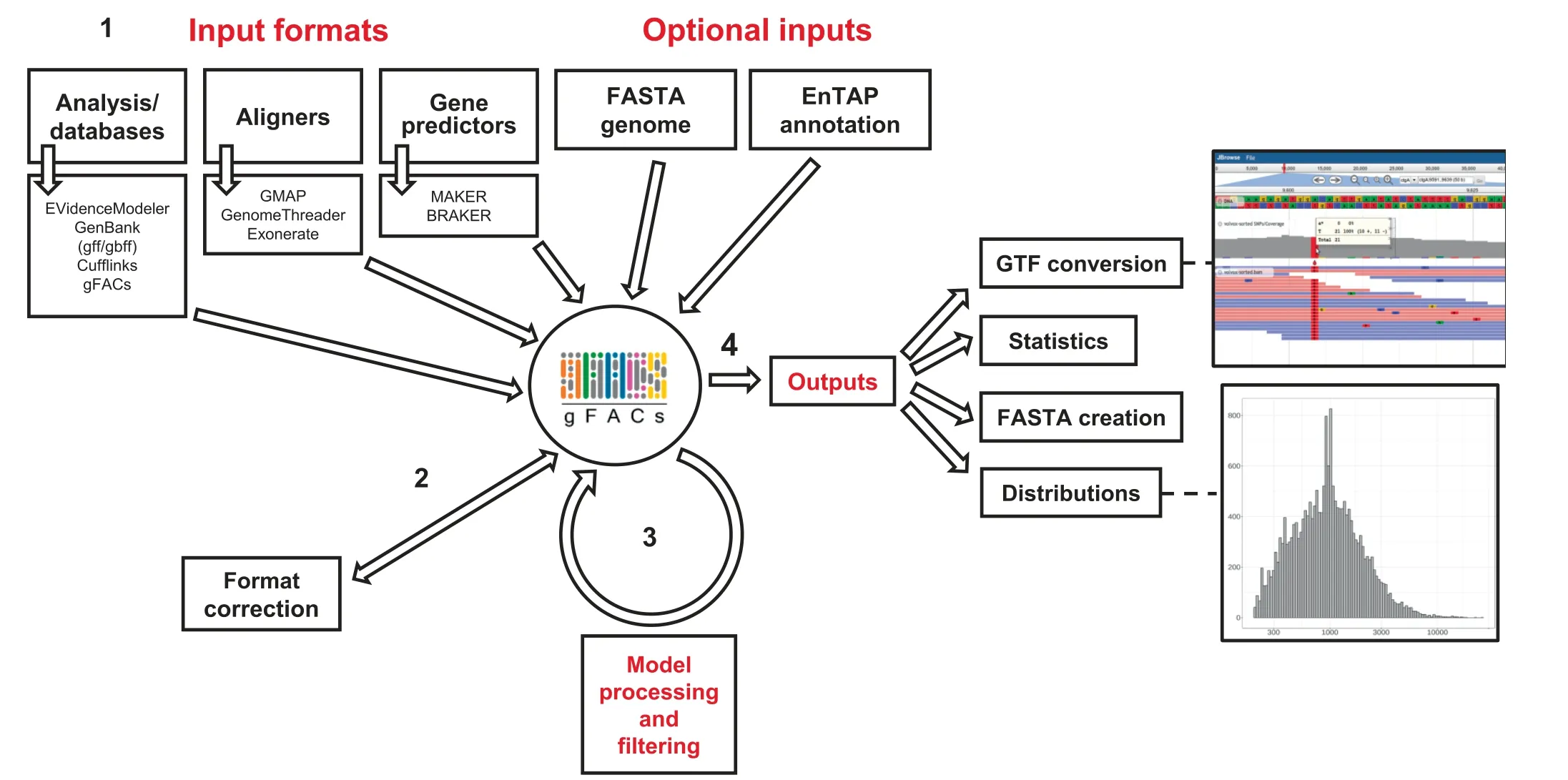
Figure 1 gFACs pipeline
gFACs removes erroneous models through a set of 14 userselected filtering options (Table 1 and Table S1), optionally aided by a reference genome or functional annotation.A notable feature of gFACs is its ability to discern and separate isoforms and conflicting gene models. This is performed by identifying overlapping exons due to conflicting evidence such as parent RNA.Splice site recognition is assumed from the provided annotation or alignment.Furthermore,gFACs is capable of collapsing these discerned isoforms and removing duplicate models to provide a final unique set. Each proposed model is subject to a predetermined set of filters as flagged by the user,many of which can be customized, such as setting minimum intron lengths or detection of in-frame stop codons. The addition of functional annotations allows for the exclusion of models without a sequence similarity search result or gene family assignment. gFACs does support alternate start codon usage, which may further increase the number of passing gene models. Alternate start sites are more common in prokaryotes(up to 20%GTG or TTG)[15]than in eukaryotes[16].Therefore,inclusion for alternate start sites is user-specified.
gFACs provides a multitude of output options in addition to a log detailing the process and filtering impacts. The primary outputs include gene/protein FASTA files, GTFrepresented models, comprehensive statistics on the selected gene models, and distribution tables. The distributions resulting from these filters can be easily imported in packages such as R to view gene lengths,CDS lengths,exon lengths,and exon size by order (Figure S1). Additionally, gFACs can generate annotation files that are compatible with SnpEff[17]for annotation of variants called against the genome and JBrowse for immediate import and visualization in a web-based genome browser [18].
Implementation
Examining protein coding gene model annotations provides insight on some of the common issues associated with annotating genomes. These can include completeness (lack of start/stop or in-frame stops),gene structure(splice sites,intron/exon lengths, and mono-exonic to multi-exonic model ratios), fragmentation (incorrect start site assignment), and lack of functional validation (similarity searches, protein domains, and gene family assignments).
To demonstrate its utility,gFACs was applied to two draft public genomes for Bos taurus (GenBank: GCF_000003055.6)[19] and Malus domestica (GenBank: GCF_000148765.1) [20]with the published annotations. BRAKER v2.1.0 annotations were generated for the model Homo sapiens and the non-model moss, Funaria hygrometrica. The human BRAKER predictions were based on RNA-seq data from two libraries (Illumina 76 bp PE, UCSC: wgEncodeEH000146). Similarly, F.hygrometrica RNA-seq data from six libraries (Illumina 150 PE, BioProject: PRJNA421369) was provided to generate the gene annotations. Microbial application was demonstrated with a Prokka v.1.11 annotation of the Borrelia burgdorferi B31, (GenBank: GCF_000008685.2) [21]. Prokka predictions are derived from a basic run on only the genome,in which candidate genes are predicted by five separate tools to generate gene coordinates. All gene models (public or generated through BRAKER/Prokka) were functionally annotated with EnTAP v0.8.0, utilizing the NCBI RefSeq database and Egg-NOG gene family database.The models generated for H.sapiens and B. burgdorferi were evaluated against the public annotation following gFACs assessments.
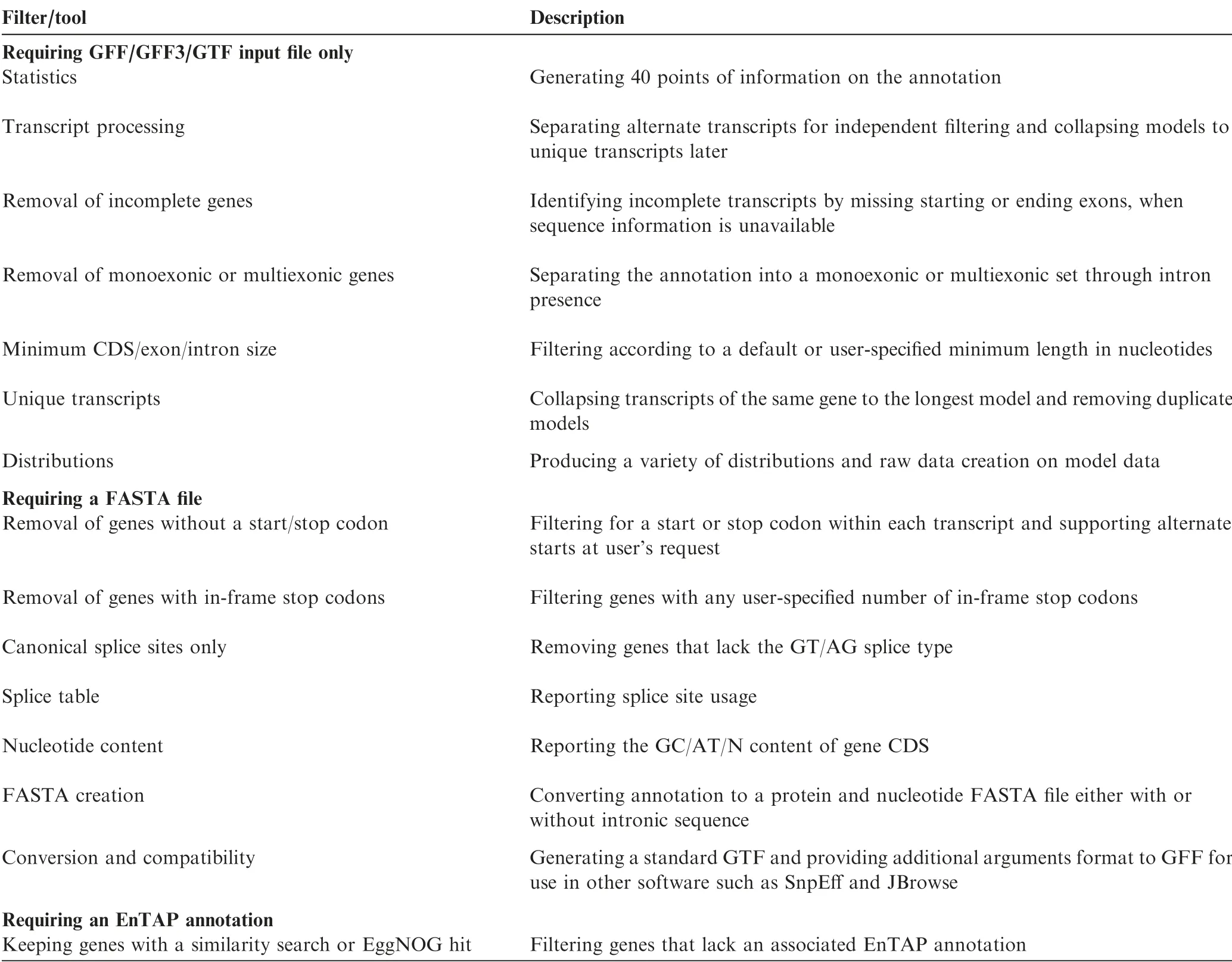
Table 1 Summary of gFACs filters and utilities (simplified)
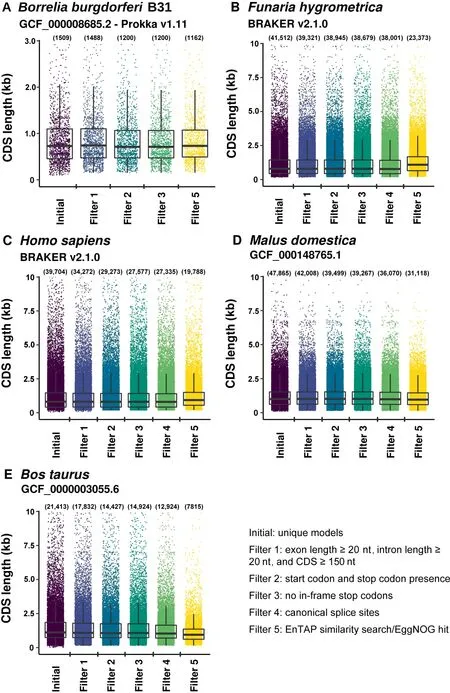
Figure 2 gFACs filtration on the annotations across five species reduces the number of gene models

Table 2 Summary of filtering and software requirements
A total of nine of the possible 14 filters were applied to the genomes representing unique sources: microbial, plant, and animal. These filters include removal of all genes with an intron or exon less than 20 nucleotides, CDS size minimum of 150 nucleotides, required presence of an ATG-only start and stop codon, no in-frame stop codons, canonical (GT/AG)only splice sites in multiexonic genes,and an EnTAP similarity search or gene family assignment. For B. burgdorferi,the canonical splice filter is not used since introns are not present.It should be noted that these filters demonstrate common issues but it would be expected that a small number of genes would have alternative splicing, micro-exons/introns, and other less common structures. These filters serve to capture clearly problematic features in these categories resulting from erroneous models.
Runtime and memory requirements vary based on the filters applied, total initial gene models, and genome size. Run specifications among the species and filters described here(Figure 2) is recorded in (Table 2).
Across all species and annotations, gFACs was able to identify gene models that were potentially problematic(Figure 2).In H.sapiens,there were 34,272 uniquely predicted genes from BRAKER, which was reduced to 19,768 using the applied filters (27,336, without considering functional annotation filter). The number of uniquely predicted genes after filtering (19,768) is comparable to the number of protein-coding genes (20,203) of the latest human genome annotation (GRCh38p.12). Detailed analysis reveals there is a larger proportion of predicted genes that match to the reference gene in the unfiltered annotation (22.4%) compared to the 19,768 genes with filtered annotation (12.5%). In terms of the match types, the proportion of perfect matches increased from 17.0%(unfiltered)to 22.1%(filtered).The proportion of perfect and contained matches increased from 26.3% (unfiltered) to 33.8% (filtered). Finally, the proportion of all types of matches increased, from 65.5% (unfiltered) to 69.3% (filtered). To further demonstrate the abilities of gFACs,a more comprehensive filtering on H.sapiens was performed (Table S1). These additional steps show the reduction in gene models in the software’s default order. Additional annotation statistics, such as splice usage, nucleotide content,and 41 statistics quantifying the final annotation are also included.
Similarly, B. burgdorferi gene predictions were reduced from 1509 to 1162 models, which represents an improvement compared to the public annotation of 1208 models(94.4%perfect matches and 98.9% total matches). Inclusion of alternative start codons in B. burgdorferi increases the number of passing models to 1416 (93.2% perfect matches and 98.1%total matches), which may include a small number of erroneous models.
The F. hygrometrica, B. taurus, and M. domestica models show similar rates of reductions through all filters including functional annotation at 43.70%, 34.99%, and 63.50%,respectively(Figure 2).The extent of model match in these species is not assessed due to the lack of a rigorous annotation.However, we noted an improvement of the models in terms of removing biologically unlikely exon, intron, and CDS lengths.
Conclusion
In summary,the gFACs software package provides a comprehensive framework for evaluating,filtering,and analyzing gene models from a range of input applications and preparing these annotations for formal publication or downstream analysis.We hope to meet the needs of the growing enthusiasm to annotate new species with a software that provides greater utility to a complex process.
Availability
gFACs is freely available and implemented in Perl with support from BioPerl libraries at https://gitlab.com/PlantGenomicsLab/gFACs.
Authors’ contributions
MC developed the software package and performed all analysis in the manuscript. MC drafted and revised with help from JW. Both authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the National Science Foundation Plant Genome Research Program of the United States (Grant No.1444573).We thank members of the Ecology&Evolution Biology Department at the University of Connecticut, including Dr.Elizabeth Jockusch,Dr.Janine Caira,Hannah Ralicki,and Dr. Kaitlin Gallagher for their valuable insight and testing. We also thank the members of the Plant Computational Genomics Lab at the University of Connecticut including Sumaira Zaman, Taylor Falk, Susan McEvoy, and Alex Trouern-Trend for their input on the software and Dr. Nasim Rahmatpour for providing the F. hygrometrica annotation.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.04.002.
 Genomics,Proteomics & Bioinformatics2019年3期
Genomics,Proteomics & Bioinformatics2019年3期
- Genomics,Proteomics & Bioinformatics的其它文章
- Diversified Application of Barcoded PLATO(PLATO-BC) Platform for Identification of Protein Interactions
- C3: Consensus Cancer Driver Gene Caller
- H3K27me3 Signal in the Cis Regulatory Elements Reveals the Differentiation Potential of Progenitors During Drosophila Neuroglial Development
- Characterization of Distinct T Cell Receptor Repertoires in Tumor and Distant Non-tumor Tissues from Lung Cancer Patients
- Warburg Effects in Cancer and Normal Proliferating Cells: Two Tales of the Same Name
- Proteomics Analysis of Lipid Droplets from the Oleaginous Alga Chromochloris zofingiensis Reveals Novel Proteins for Lipid Metabolism