
Integrating Culture-based Antibiotic Resistance Prof ileswith Whole-genome Sequencing DatAfoR11,087 Clinical Isolates
2019-07-12 06:35:20ValentinAGalataCedricLacznyChristinABackesGeorgHemMrichStanisakSusanneSchmolkeAndreFrankeEckartMeeseMathiasHerrmannLutzvonMullerAchiMPluMRolfMullerCordStahlerAndreasPoschAndreasKeller
ValentinAGalata,Ce′dric C.Laczny,ChristinABackes,Georg HemMrich-Stanisak ,Susanne Schmolke,Andre Franke,Eckart Meese,Mathias Herrmann,Lutz von Mu¨ller,AchiMPluM,RolfMu¨ller,Cord Sta¨hler,Andreas E.Posch*,Andreas Keller*
1 ChaiRfoRClinical Bioinformatics,Saarland University,66123 Saarbru¨cken,Germany
2 Institute of Clinical MoleculaRBiology,Christian-Albrechts University of Kiel,24105 Kiel,Germany
3 Siemens Healthcare GmbH,Strategy and Innovation,91052 Erlangen,Germany
4 Department of Human Genetics,Saarland University,66421 Homburg,Germany
5 Institute of MedicalMicrobiology and Hygiene,Saarland University,66421 Homburg,Germany
6 Ares Genetics GmbH,1030 Vienna,Austria
7 Curetis GmbH,71088 Holzgerlingen,Germany
8 Department of Pharmacy,Pharmaceutical Biotechnology,Saarland University,66123 Saarbru¨cken,Germany
9 Department of Microbial Natural Products,Helmholtz-Institute foRPharmaceutical Research Saarland(H IPS),Saarland University,66123 Saarbru¨cken,Germany
10 Helmholtz CenteRfoRInfection Research and Pharmaceutical Biotechnology(HZI),Saarland University,66123 Saarbru¨cken,Germany
KEYWORDS Antibiotic resistance;Who le-genoMe sequencing;Bacteria;Pan-genoMe
Abstract Emerging antibiotic resistance isamajoRglobalhealth threat. The analysisof nucleic acid sequences linked to susceptibility phenotypes facilitates The study of genetic antibiotic resistance deterMinants to inforMmoleculaRdiagnostics and drug development.We collected genetic data(11,087 neWly-sequenced whole genomes)and culture-based resistance prof iles(10,991 out of The 11,087 isolates coMprehensively tested against 22 antibiotics in total)of clinical isolates including 18 main species spanning Atime period of 30 years.Species and drug specific resistance patterns were observed including increased resistance rates foRAcinetobacteRbaumannii to carbapeneMs and foREscherichiAcoli to fluoroquinolones.Species-level pan-genomes were constructed to reflect The genetic repertoire of The respective species,including conserved essentialgenesand known resistance factors.Integrating phenotypes and genotypes through species-level pan-genomes allowed to infeRgene-drug resistance associations using statistical testing.The isolate collection and The analysis results have been integrated into GEAR-base,Aresource available foRacadeMic research use free of charge at https://gear-base.com.
Introduction
The development of neWantiMicrobial drugs has largely stagnated oveRThe last feWdecades[1],while The drug resistance ratesof many pathogenshaveat The same timebeen increasing[2-4].Various large-scale efforts have been launched to investigate The emerging drug resistance,such as The MeropeneMYearly Susceptibility Test Information Collection(MYSTIC)program[2],The Canadian National Intensive Care Unit(CAN-ICU)study[5],The Canadian National Surveillance(CANWARD)study[6,7],The CenteRfoRD isease DynaMics,EconoMics and Policy(CDDEP)study[3],and The European AntiMicrobial Resistance Surveillance Network(EARS-Net)survey[8].The results of The se studies have shed light on The most common bacterialpathogensand resistance rates foRregularly adMinistered antibiotics,With The primary focus on The trend analysis of specific bacterial groups,periods of time,oRlocations[2,3,9-12].The global challenge of emerging drug resistance is fur The Rexacerbated by The rising prevalence of MicroorganisMsWith multidrug resistance(MDR)phenotypes[13].Accordingly,identifying and adMinistering The most effective drug in each individual case is of even greateRiMportance foRsuccessful treatmentof bacterial infections.However, The se studies did not investigate The genetic repertoire of The pathogens,which represents an important source of information—e.g.,The resistance genotype may be readily revealed while The respective phenotype isMisleading oRnot expressed undeRartificial laboratory conditions[14,15].
Simultaneously,The recovery of genoMic information froMMicroorganisMs viAhigh-throughput sequencing approaches has become Aroutine task.This not only allows The highresolution study of individual organisms’genomes,but also The aggregated study in The forMof ‘‘pan-genomes”—The united genetic repertoire of Aclade[16].Pan-genomes can be used to identify common genetic potential—i.e., The ‘‘core”genes of Aclade—as well as genes that are less broadly conserved(‘‘a(chǎn)ccessory”or‘‘singleton”genes)[16].This facilitates The identification of essential genes oRgenes that provide adaptation advantages.Multiple coMputationalapproachesareavailable foRThe systematic creation of pan-genomes,e.g.,Roary[17],EDGAR[18],and panX[19].AsAresult,Avariety of bacterial pan-genomes,typically at The species-level,have thus faRbeen constructed[20-23].However,most pan-genome studies focuson distinct speciesand do not always coveRclinically relevantspecies.FoRexaMple,MetaRefrepresentsAresource that provides information about pan-genomes froMmultip le species and integrates approximately 2800 public genomes[24].Although The diversity of The The rein included organisMs is particularly broad,The depth is liMited in relation to clinically relevant bacteria—e.g.,seven K lebsiellApneumoniae genomes.Moreover,individual isolates included in The studies of ten span narroWtime frames and/oRhave liMited geographic spread.
While pan-genoMic studies typically focus on The genetic information alone,efforts combining genoMic and phenotypic information,in particulaRfroMantibiotic resistance testing,foRThe study of conserved oRemerging resistancemechanisms are becoMing increasingly prevalent[25-28]. The re aremany antibiotic resistance resources available[29],howeveRonly feWlink genoMic and phenotypic information of bacterial isolates.One of such resources is The PathosysteMs Resource Integration Center(PATRIC)[30],which represents Arich service foRThe study of >80,000 genomes[31].Yet,antiMicrobial resistance information is available only foRabout 10%of The genomes.Fur The rmore,as The genomes and The associated metadatAof PATRIC are iMported froMpublic resources,which are populated by individual research efforts,datAstandardization oRnormalization is challenging.Finally,individual taxAmay be underrepresented and thus warrant expansion—e.g.,The numbeRof EscherichiAspp.genomesWith antiMicrobial resistance metadatAis almost two orders of magnitude smalleRthan that of MycobacteriuMspp.genomes[31].
Motivated by The importance of linking resistance phenotypes With genoMic features,we collected whole-genome sequencing datAof 11,087 clinical isolates representing,inteRalia,18 main bacterial species.The saMp les were collected in North America,Europe,Japan,and AustraliAoveRAperiod of 30 years,and processed in Aconcerted effort, The reby reducing experimental bias.Culture-based resistance testing was performed foR10,991 out of The 11,087 isolates against 22 antibiotic drugs.Fur The rmore,species-level pan-genomeswere constructed on The basis of per-isolate de novo assemblies and were used to infeRgene-drug resistance associations.This wealth of information is integrated into an online resource,Genetic Antibiotic Resistance resource, or in short,GEAR-base(Figure 1).Providing broad organismal,antibiotic treatment and temporal coverage,GEAR-base is expected to support The pan-genome-based study of bacteriAand to advance research on known oRemerging antibiotic resistance mechanisms.GEAR-base is available foRacadeMic research use free of charge at https://gear-base.com.
Results
Resistance testing of cultured bacterial isolates
The present dataset of 11,087 bacterial isolates covered Atotal of 6 faMilies,14 genera,and 20 species(considering species With at least50 isolates,Table S1)and comprised two datasets:1001 isolates froMThe Staphylococcus aureus strain collection and 10,086 isolates froMThe G ram-negative collection.FroMThe S.aureus strain collection,993 isolates were tested foRmethicillin resistance and susceptibility(see Methods section).FoR9998 isolates froMThe G ram-negative collection,culturebased antiMicrobial susceptibility testing (AST)foR21 commonly-prescribed Food and D rug AdMinistration(FDA)-approved antibiotics from 8 drug classes was performed to deterMine The respective MinimuMinhibitory concentrations(MICs)(Figure 2A).The resistance prof iles were deterMined foReach isolate in accordance With The European ComMittee on AntiMicrobial Susceptibility Testing(EUCAST)guidelines(v.4.0)foRAtotal of 182 drug concentrations(7-11 concentrations peRdrug;Tables S2 and S3,Figure 2B). Whole-genome sequencing (WGS)-based taxonoMic identification was performed foRall isolates[32].In The folloWing content,we focused on The analysis results of The MICs and resistance prof iles of The 9998 isolates froMThe G ram-negative collection.
All patient-derived isolateswere collected in clinics located in North America,Europe,Japan,oRAustraliAfroM1983 to 2013(Figure S1).Varying degrees of resistancewere observed among The isolates(Figure 2B).The majority of species demonstrated relatively low resistance rates(<20%)to aMinoglycosides(gentaMicin and tobraMycin)and carbapenems(ertapenem,iMipenem,and meropenem),except foRAcinetobacteRbaumannii(≥29%foRaMinoglycosides and meropenem),Pseudomonas aeruginosa(26%foRgentaMicin),and K lebsiellApneumoniae(26%foRtobraMycin). The se rates were coMpared against two independent large-scale studies—CDDEP (USA-based results;CDDEP ResistanceMap,https://resistanceMap.cddep.org/AntibioticResistance.php,accessed on SeptembeR26,2017)[3]prograMand The MYSTIC program[2],formatching species and drug data.Both studies report low(<20%)resistance rates foRThe aMinoglycosides and carbapenems during The observation period (1999-2012/2014 foRCDDEP and 1999-2007/2008 foRMYSTIC)except foRA.baumannii(CDDEP:>20%since 2005 foRcarbapenems and>35%during 1999-2012 foRaMinoglycosides;MYSTIC:>37%in 2007/2008 foRcarbapeneMs and>20%during most years foRaMinoglycosides).FoRK.pneumoniae and tobraMycin(aMinoglycosides foRCDDEP),MYSTIC and CDDEP reported>10%resistance rates since 2005 With only one value of above 20%observed by MYSTIC in 2007.Finally,foRP.aeruginosAand gentaMicin,MYSTIC reported Aresistance rate of only around 10%.The rate of isolates resistant to multiple antibiotic drugs,i.e.,resistant to at least three drugs froMdifferent drug classes(CDDEP ResistanceMap),was highest foRA.baumannii(44%)and foREnterobacteRspp.(41%-45%).FoRThe remaining species and drug classes, The MDRrateswereat least20%,except foRAcinetobacteRcalcoaceticus(0%),SalmonellAenterica(11%),and ShigellAspp.(0%-3%).In addition to The investigation of individual species-drug combinations,we analyzed whe The Rdrug pairs showed correlating MIC prof ilesamong allisolates(Figure S2).In general,The highest correlations were expected ly found within separate drug classes—e.g.,foRfluoroquinolones,aMinoglycosides,and carbapeneMs.While foRsome species,e.g.,BurkholderiAcenocepacia,AcleaRclustering according to drug classes and The iRmechanisMof action was observed,o The Rspecies,such as S.enterica,showed lesspronounced clusteRstructures.
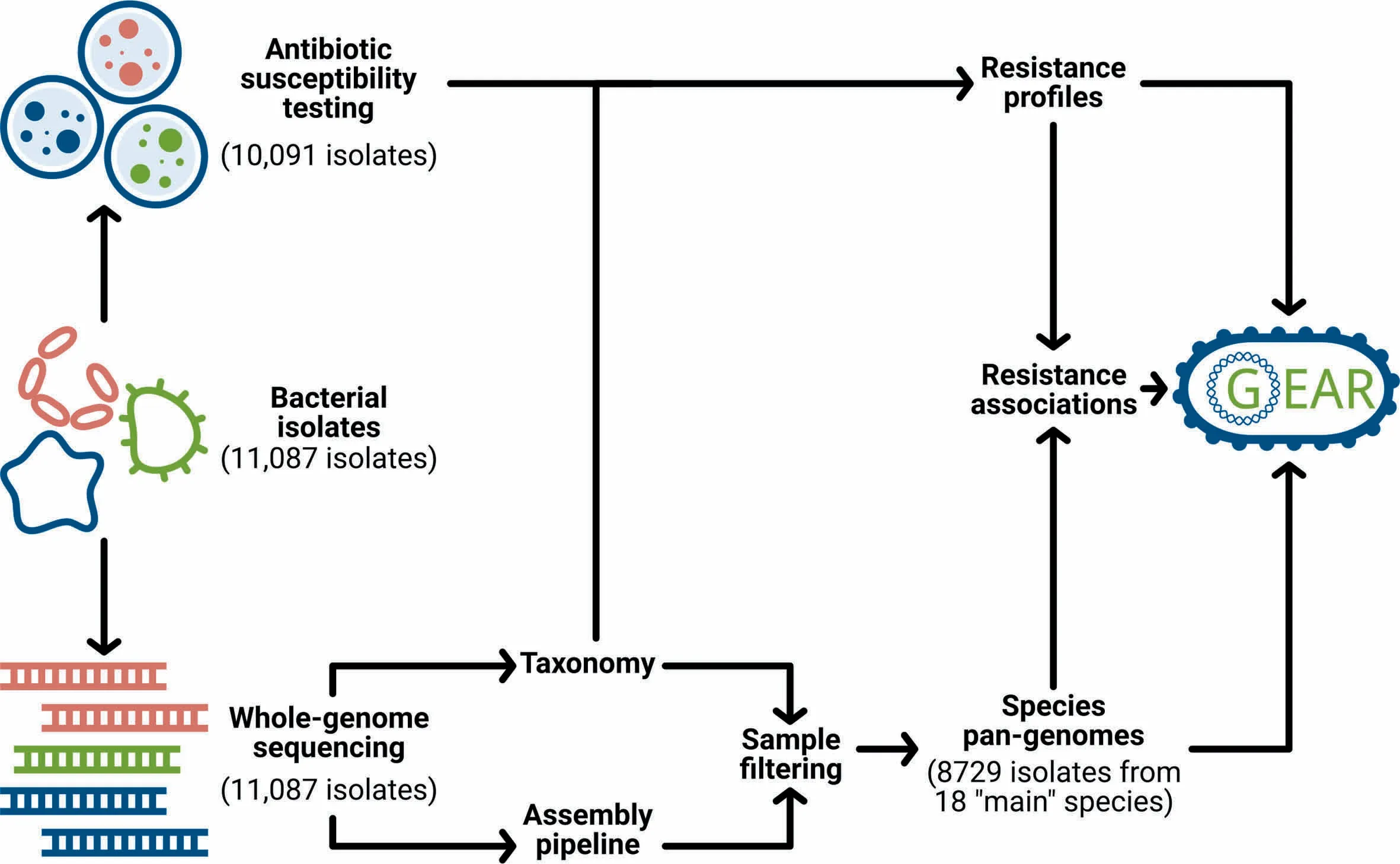
Figure 1 GEAR-basework floWand structureSchematic overvieWof datAcollection,processing and integration into GEAR-base.
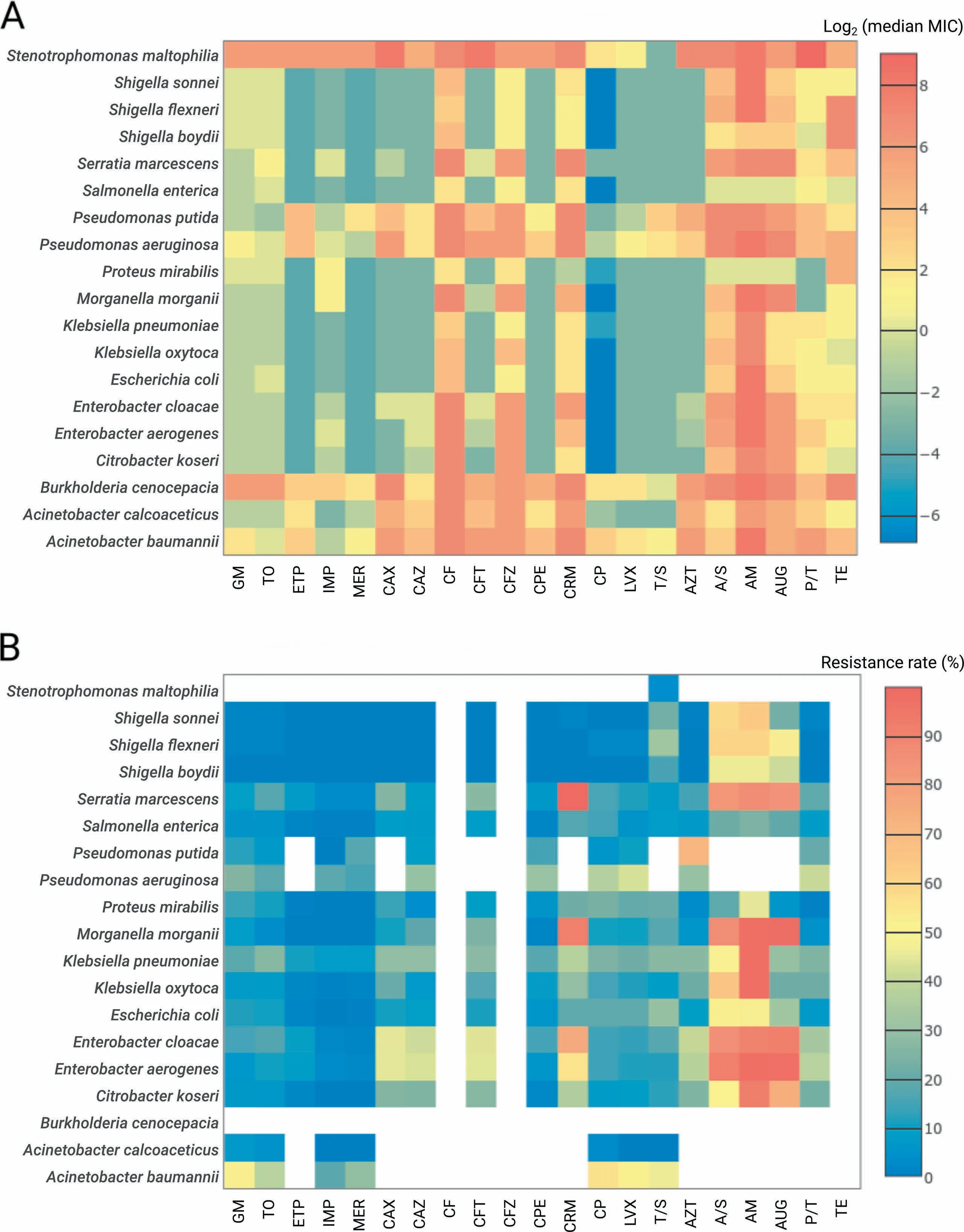
Figure 2 OvervieWof resistance prof ilesHeatmapsof log-transformed(base2)median MIC values(A)and resistance rates(B)foRallspeciesWith at least 50 isolates.D rugs labels were grouped relative to The iRclass.The cells are coded in coloRgradient froMblue to red With blue foRloweRvalues and red foRhigheRvalues.White coloRin panel B corresponds to The cases where no breakpoints are available froMThe used guidelines.MIC,MinimuMinhibitory concentration.
Subsequently,we compared resistant and non-resistant isolates With respect to The iRcollection yeaRin ordeRto identify potential trends of de-/increasing antibiotic resistance rates(Figures S3 and S4,and Table S4).The folloWing species-drug pairswere found to exhibit particularly loWP values[WMWtest,false discovery rate(FDR)ad justed P<1E-17],aswell as increases in resistanceoveRtime:K.pneumoniae to cefepime,
K.pneumoniae and A.baumannii to carbapenems,and E.coli to fluoroquinolones.SiMilaRtrendswere reported by The CDDEP[3]program(CDDEP ResistanceMap)and The MYSTIC program[2],including increasing resistance rates foRA.baumannii to carbapenems(43%froM1999 to 2014 in The USA,CDEEP),and foRE.coli to fluoroquinolones(30%froM1999 to 2014 in The USA,CDEEP;>20%froM1999 to 2008,MYSTIC).
While The culture-based analyses provide species-resolved information about resistance rates oveRtime and corroborate previous findingson The global increase in antibiotic resistance,genetic features represent important factorsand were thus concoMitantly considered.
Whole-genome de novo assembly of isolates and species pangenomes
A total of 11,087 bacterial isolates were whole-genome sequenced using IlluMinAH iseq2000/2500 sequencers,resulting in amedian numbeRof 1,517,147 paired reads peRisolate(1,609,533±620,481).De novo assemblies were successfully created foR11,062(99.8%)isolates(Figure 3)and of The se,The assembled genomes of 10,764(97.3%)isolates passed The stringent assembly quality criteria.Moreover,The assembled genomes of 9206(83%of 11,087)isolates fulfilled The quality criteriAfoRtaxonoMic assignment.Atotal of 8729 isolates,representing 18 main species having≥50 isolates,were used afteRstringent quality filtering(see Methods foRsaMple filtering details)in The subsequent analyses and in The construction of species-level pan-genomes(Table S3).

Figure 3 Assembly quality overviewAssembly summary statistics foR The 11,062 isolatesWith Adenovo assembly.The isolatesweregrouped by The iRspecies taxon,and isolates not belonging to any of The main 18 species used foRpan-genome construction were grouped into”O(jiān) The r”.The box plots shoWThe GC content(A),mean assembly coverage(B),numbeRof contigs(C),L50 value(D),and N 50 value(E)foRcontigs of at least 200 bp.The assembly quality cut of fvaluesare illustrated by dotted lines(1000 foRThe numbeRof contigs;200 foRL50;and 5000 bp foRN 50).The p lot areAsatisfying The respective filtering criterion is colored in green.Percentages of isolates passing The respective criterion aswell as all criteriAare shown to The right.
First,The presence/frequency of genes froMAset of 111 single-copy markeRgenes,which were defined as essential genes by Dupont et al.[33],was used as Aproxy to estimate The genome comp leteness of individual de novo assemblies.Overall,The assemblies were found to be largely comp lete.92 essential genes(82.9%)were identified in at least 99%of The 8729 isolates(Figure S5)thatwere used to construct Aphylogenetic tree of The se isolates(Figure S6).Fur The rmore,speciesspecific presence/absence patterns were frequently observed(Figure S7A).FoRexample,TIGR00389(glycine-tRNAligase)was only found in S.aureus,whereas TIGR00388(glycine-tRNAligase,alphAsubunit)was not present in this species.FouRgenes,TIGR00408(encoding The proline-tRNAligase),TIGR02387(encoding The DNA-directed RNApolymerase,gammAsubunit),TIGR00471(encoding The phenylalanine-tRNA ligase,beta subunit),and TIGR00775(encoding The Na+/H+antiporter,NhaD faMily),were not found in any of The isolates,except foRsporadic hits in Pseudomonas aeruginosAfoRTIGR00408.
In The next step,Resfams core-based resistance factors[34]were annotated in The isolate assemblies in ordeRto study The species-leveldistribution of The segenetic features. The numbeRof covered Resfams(mean count of hits≥1)varied between species froM4.1%(5 of 123 ResfaMs,MorganellAmorganii)to 11.4%(14 of 123 Resfams,A.baumannii and ShigellAsonnei)(Figure S8).Three ResfaMs were found in at least 90%of all considered isolates. The se are allantibiotic efflux puMps,which include RF0007[ATP-binding cassette(ABC)type],RF0107(ABC type),and RF0115[resistance-nodulation-cell division(RND)type],With The latteRhaving amean count of hits of ≥5 foR14 out of 18 species.
The multi-locus sequence typing(MLST)analysis revealed,that in all species With Atyping scheme included in The used version of PubMLST,isolateswereassigned to at least6 different sequence types(STs),except foRS.sonnei,and neWSTs could be identified,except foRShigellAflexneri and S.sonnei(Figure S9).Among The se species,The proportion of isolates Without Aconfident assignmentwas high(≥10%)foRB.cenocepacia,EnterobacteRcloacae,KlebsiellAoxytoca,and Stenotrophomonasmaltophilia.
The sizeof The speciespan-genomes(i.e., The numbeRof centroids)ranged froM5838(S.aureus,total pan-genome length<5Mb)to 42,046(E.cloacae,total pan-genome length>30Mb)(FigureS10).Acentroid refershere to The representative gene of Ahomologous gene clusteRWith≥90%pair-Wise aMino acid sequence identity(Methods).Most centroidswere found in<10%oRin≥90%of The isolates(Figure4).Moreover,allpan-genomeswere found to beopen based on The analysisof The numbeRof centroids in relation to The numbeRof included genomes(Figure S11,Table S5).The two-dimensionalembedding of The core centroids froM The pan-genomes revealedmany taxon-specific patterns(Figure S12)With distinct clusters foRB.cenocepacia,M.morganii,A.baumannii,Proteusmirabilis,S.aureus,S.maltophilia,P.aeruginosa,and Serratiamarcescens.We coMpared The numbeRof (core)centroids in ouRpangenomes to The numbers reported by panX[19](http://pangenome.tuebingen.mpg.de,accessed on January 29,2018).The numberof centroidspresent in at least90%of The analyzed genomeswas consistent foRallmatching species(Table S6).However,The pan-genome size,i.e.,The total numbeRof centroids described in GEAR-Base,was siMilaRfoRE.coli and S.aureus,butexceeded substantially The numberof centroidsdescribed in panX foRA.baumannii,K.pneumoniae,P.aeruginosa,and S.enterica(Table S6).With respect to The presence of essential genesin The species-levelpan-genomes, The mean numberof centroids containing at least onematching genewas one,that is, The se essential genes weremostly found in only one centroid cluster(Figure S7B).However, The mean numbeRof centroids was≥1.25 foReight essential genes,i.e.,in some species The se geneswere found inmultip le centroid clusters.
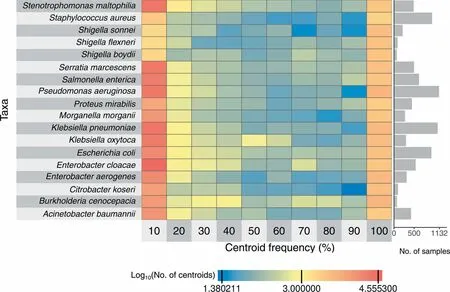
Figure 4 Centroid frequencyNumbeRof centroids in each pan-genomeof The 18main species in relation to The iRfrequency.The first column contains centroids thatare present in<10%of The isolates,and The last one contains centroids that are present in≥90%of The isolates.Cells are coded in coloRgradient to indicate The log10-transformed numbeRof centroids.The baRp loton The right shows The numbeRof isolatesused to construct The respective pan-genomes.
In The folloWing section,The resistance phenotypes and genoMic features were linked and significantly associated centroids were fur The Rstudied,With respect to The iRoverlap to known resistance genes froMThe Resfams core database.
Resistance associations by linking phenotype and genotype
We used binary information in The forMof centroid presence/absence to test foRsignificant centroid-drug associations peRspecies.The numbeRof found associations ranged froMbeloW10 to above 500;most associations(≥500)were found foRP.aeruginosAand tobraMycin,and K.pneumoniae and gentamicin(Figure 5).Fur The rmore,The drug resistance-associated centroids encoding foRAresistance gene were investigated.FroMThe Resfams core database,45 of The 123 factors were found in at least one centroid(Figure S13).Among The se,The top ten Resfams genes froMboth analyses covered various resistance mechanism classes - nucleotidyltransferases,phosphotransferases,acetyltransferases,beta-lactamases,and major facilitator superfaMily (MFS) transporters(Figure S13B).
GEAR-base online resource
The GEAR-base resource is freely accessible at https://gearbase.coMfoRacadeMic research use and currently provides two modules foRbrowsing of The database—Aculture-based module and Apan-genomemodule—as well as Amodule foRThe analysis of user-provided data.The culture-based module is focused on The G ram-negative isolate collection and provides an interactive view of The taxonoMic coMposition,MIC,and resistance prof iles,as well as additionalmeta-data,e.g.,collection yeaRoRisolate distributions.The pan-genome module provides general statistics,such as assembly quality of The included isolates,pan-genome size,and resistance association analysis overview,foRboth The G ram-negative and The S.aureus isolates.Gene nucleotide sequences can be downloaded foReach individual pan-genome centroid and Abatch-download of all centroid nucleotide sequences is available.Moreover,pan-genome centroids can be browsed online foRspecific gene products and filtered by The iRpresence in The isolates.In addition,centroid clusters can be viewed including associated gene annotations, The hits to The Resfams core database,and information about potential resistance associations against The setof herein included drugs.GEAR-base’sanalysis module allows The useRto query individual gene sequences against The pan-genome centroid sequences using Sourmash[35],against hidden Markov models(HMMs)of pan-genome centroid clusters and ResfaMs core database using HMMER(http://hmmer.org/),and against The NCBI nt/nRdatabase using BLASTp[36].Fur The rmore,Agenome-scale search against The present clinical isolate collection,The finished genomes froMThe NCBI RefSeq database[37],as well as The National Collection of Type Cultures(NCTC)3000 genomes project froMThe Public Health England and The Wellcome Trust Sanger Institute (http://www.sanger.ac.uk/resources/downloads/bacteria/nctc/,accessed on OctobeR18,2017)can be performed online using Mash/MinHash[38].
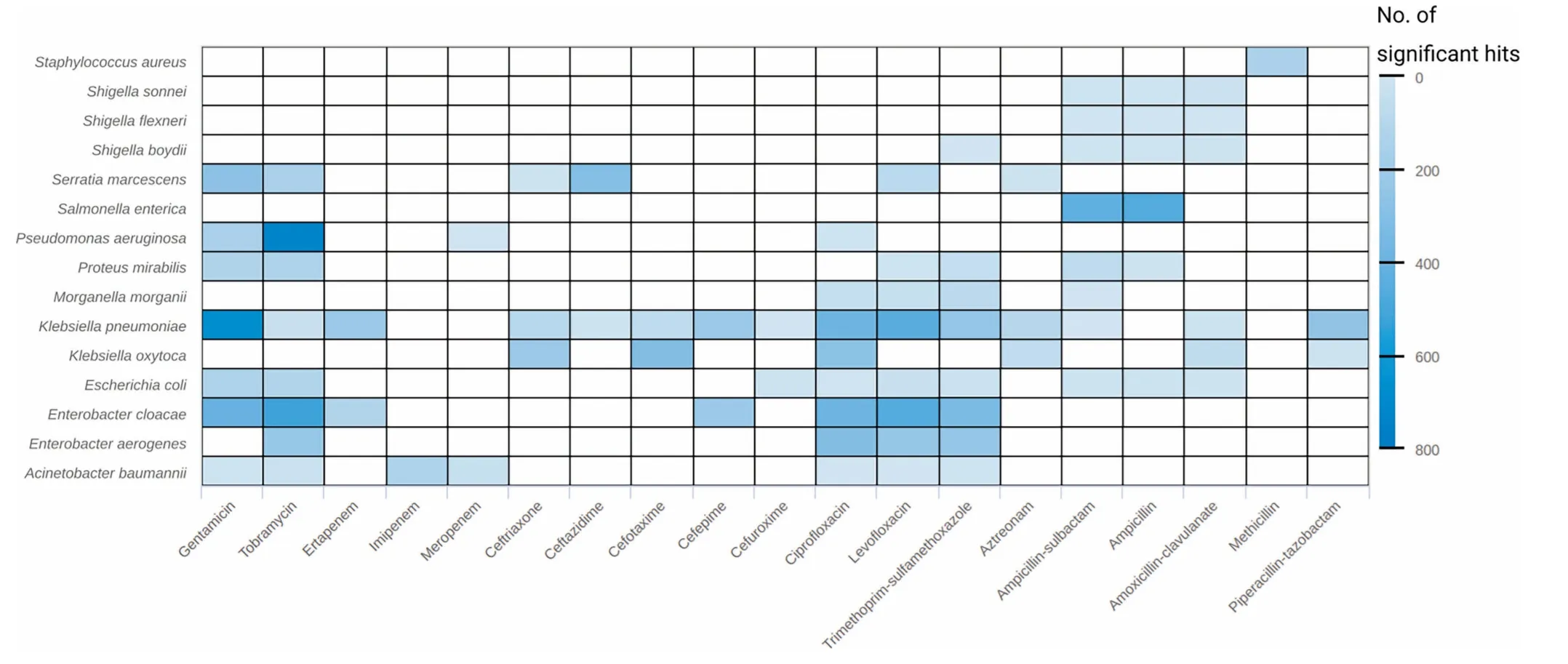
Figure 5 NumbeRof significant results of The resistance association analysisSignificant results(adjusted P<1E-5)of The resistance association analysis based on The presence/absence of centroids.The heatmap shows The numbeRof significant results(in coloRgradientwith lighteRblue foRsmalleRnumbers and darkeRblue foRlargeRnumbers)peRtaxon and drug.D rugs are sorted according to The iRclass.
We used Arecently-published K.pneumoniae genome[39](strain 1756, NCBI assembly accession ID GCF_001952835.1_ASM195283v1)to demonstrate The analysis functionalitiesof GEAR-base.In Afirst step,The chromosome and p lasMid sequences were up loaded and Aperfect match was found to The genome’s NCBI entry,as expected.The nextbestmatcheswere to AK.pneumoniae isolate froMThe current collection of clinical isolates(828/1000 shared hashes,distance of 4.71E-3),and to AK lebsiellAsp.genome(ERS706555)froMThe NCTC 3000 database(709/1000 shared hashes,distanceof 8.89 E-3).In Asecond step,all coding DNAsequences(CDS)were searched against The pan-genome centroids in GEARbase using Sourmash and against The Resfams core database. The majority of The pan-genome hitswere related to K.pneumoniae(6206 hits of 11,267)followed by E.aerogenes(1537 hits)and K.oxytoca(1014 hits).S.aureus,AG ram-positive species,served as an outgroup and no hits to its pan-genome were found.In total,37 hits to 21 unique ResfaMs(core database)were found in The query genome CDSWith 23 hitson The chromosomeand 14 on The p lasMid.The top threemostoccurring Resfams were RF0115(8 hits,RND antibiotic efflux puMp),RF0098(3 hits,multidrug efflux RND membrane fusion protein MexE,RND antibiotic efflux),and RF0053(3 hits,class Abeta-lactamase).Fur The rmore,The CDSs of eight antibiotic resistance genes reported in The original genome announcement were investigated.The HMM-based search of pan-genome centroids resulted in The identification of two chromosomal CDSs,WP_076027158.1(multidrug efflux RND transporteRperiplasMic adaptoRsubunit OqxA)and WP_004146118.1(FosAfaMily fosfoMycin resistance glutathione transferase),being classified as K.pneumoniae-derived centroids according to The iRtop hits(with respect to The full sequence score). The top hits of The remaining genes(5 plasMid-derived and 1 chromosome-derived)included centroids froMo The RG ram-negative species.However,The centroid clusteRannotations matched The expected protein functions foRall eight CDSs independent of The species.The top three hits foRWP_004146118.1 were centroids from
K.pneumoniae,E.aerogenes,and K.oxytoca,matching The expected annotation and present in almost all isolates(>98%)of The respective pan-genomes.This high prevalence matches The observationsmade by RyotAet al.reporting similarly high frequency(>96%)of fosAin The se species[40].FoRThe beta-lactamasesWP_004176269.1(class Abroad-spectruMbeta-lactamase SHV-11)and WP_000027057.1(class AbroadspectruMbeta-lactamase TEM-1),The top hits in K lebsiellAwere associated With resistance to penicillins and cephalosporins.And foRThe aMinoglycoside transferases WP_000018329.1 (aMinoglycoside O-phosphotransferase APH(3′)-Ia),WP_032491824.1(ANT(3′′)-IAfaMily aMinoglycoside nucleotidyltransferase AadA22),and WP_000557454.1(aMinoglycoside N-acetyltransferase AAC(3)-IId),The top hits in K.pneumoniae were associated With resistance to aMinoglycosides.Moreover,all three chromosome-derived CDSs(WP_004176269.1,WP_076027158.1,and WP_004146118.1)matched to centroids found in>92%of The K.pneumoniae isolates, two of The five p lasMid-derived CDSs(WP_032491824.1 andWP_000027057.1)matched to centroids with Afrequency of >25%,while The remaining CDSs matched to centroidsWith Afrequency of <12%.
Discussion
To facilitate The studies on antibiotic resistance,we have built GEAR-base,Aresource incorporating paired datAon resistance phenotypes and genoMic features foRan extensive,longitudinal collection of clinical isolates froMvarious bacterial species.This concerted effort is expected to reduce experimentalbiasand The present resource providesAportal foRinformation retrieval aswell as datAanalysis.
Species-level antibiotic resistance phenotypes can be inspected using The culture-based module in GEAR-base.Specifically,resistance rates and trends acrossmultiple species and antibiotic drugs can be assessed on Alarge scale,which we believe is important foRcurrent and future antibiotic resistance research.Although some effect of potential saMpling bias cannot be excluded,ouRfindings on The increased resistance rates corroborate previously reported trends.In addition to this phenotypic information,genoMic information is included in The pan-genome module.Such information can be used independent of The phenotypic information,i.e.,purely froMApan-genoMic perspective,e.g.,foRThe study of inter-oRintra-species gene conservation.The observed numbeRof core centroids was consistent With The statistics reported by panX.However,GEAR-base pan-genomes are based on significantly higheRsaMple numbeRand are substantially largeRin size,thus giving access to AcoMprehensive collection of The genome heterogeneity foRhuman bacterial pathogens.In addition,GEAR-base links The se two information layers through centroid-drug associations. The se associations can subsequently be exp lored to study resistance mechanisMs.Fur The rmore,individual researchers can compare genes oRgenomes of interest to The present resource, The reby providing an independent layeRof support.This functionality was demonstrated using a recently published carbapenem-resistant K.pneumoniae isolate.While The taxonoMic classifications of The genome and of a set of chromosome-derived antibiotic resistance genes are consistent With The expected taxonoMy of The isolate,The p lasMidderived antibiotic resistance genes exhibit ambiguous taxonoMic assignments,which is not unexpected foRp lasMidborne genes.Moreover,The extensive collection of isolates included herein enables The study of The overall conservation degrees and The time-resolved frequencies of this exeMp lary antibiotic resistance gene set.
The analysis functionality in GEAR-base covers external genome databases(NCBIRefSeq as well as The NCTC 3000 genomes project froMPublic Health England and The Wellcome Trust SangeRInstitute)in addition to The present collection of clinical isolate genomes.However,because The majority of external genomes are not linked to antibiotic resistance information and centroid-drug associations are considered Akey component of The present resource,The pan-genomemodule is restricted to The present isolates.Additionally,The species-level pan-genome centroids in GEAR-base are available foRdownload and provide Agreat opportunity foRsubsequent integration With external genomes foRfur The Rstudy.
Emerging antibiotic resistance represents a multidiscip linary and global challenge.We believe that GEARbase Will serve as Avaluable resource enabling The detailed analysis of resistance-associated genoMic features.GEARbase includesAcomprehensive selection of clinically highly relevanthumanMicrobialpathogensand Will thusbe of great use foRThe research and clinical communities.
Materials andmethods
Bacterial isolates
The dataset of 11,087 isolates consisted of 1001 isolates froMThe S.aureus strain collection of Saarland University Medical CenteRand Acollection of 10,086G ram-negativebacterialclinical isolates that forMpart of The Microbiology strain collection of Siemens Healthcare D iagnostics(West Sacramento,CA)[32].DNAextraction using The Siemens VERSANT?samp le preparation system [41]and whole-genome nextgeneration sequencing were performed foRall isolates as described in GalatAet al.[32](2×100 bp paired-end on IlluMinAH iseq2000/2500 sequencers).
Methicillin susceptibility of S.aureus isolates
FoR993 isolates froMThe S.aureus strain collection,detection of methicillin-resistant oRmethicillin-susceptible Staphylococcus aureus(MRSA/MSSA)isolates was performed.The specimen were p lated on CHROMagaRMRSAdetection bip lates(Mast,Reinfeld,Germany).All MRSA-positive culture isolates were fur The Rconfirmed using a penicillinbinding protein 2Alatex agglutination test(Alere,Ko¨ln,Germany).
Susceptibility testing and resistance prof iles of Gram-negative isolates
FoR9998 isolates froMThe G ram-negative isolate collection,AST was performed.Frozen reference AST panelswere prepared folloWing Clinical Laboratory Standards Institute(CLSI)recommendations[42]. The antiMicrobial agents included in The panels are provided in Table S2.PrioRto use With clinical isolates,AST panels were tested and considered acceptable foRtestingWith clinical isolateswhen The QC results met QC ranges described by CLSI[42].
Isolateswere cultured on trypticase soy agarWith 5%sheep blood(Be The sdABiological Laboratories,Cockeysville,MD)and incubated in ambient aiRat 35±1°C foR18-24 h.Isolated colony panelswere inoculated according to The CLSI recommendations(CLSI additional reference)and incubated in ambient aiRat 35±1°C foR16-20 h.Panel resultswere read visually,and MICswere deterMined.
MIC value processing
The bacterialculturemay notgroWfoRThe lowestdrug concentration tested(expressed as≤x)oRshoWno significant groWth decrease foRThe highest concentration tested(expressed as>x),where x represents The drug concentration tested.To alloWconsistent processing, The se MIC values were transformed as follows:in The formeRcase,The MIC value was set to be x/2(e.g.,‘‘≤0.25”was set to‘‘0.125”),and in The latteRcase,The MIC value was set to be x*2(e.g.,‘‘>64”was set to‘‘128”).Additionally,we considered only The MIC value of The first agent in case of drug combinations(e.g.,‘‘32/16”was set to‘‘32”).
D rug information
The 21 drugs used in this study were grouped into 8 drug classes based on The iRcategory in The EUCAST guidelines[43].Among The m,7 drugsbelong to cephalosporins(cefazolin and cephalotin-1st generation;cefuroxime-2nd generation;cefotaxime,ceftazidime,and ceftriaxone-3rd generation;and cefepime-4th generation),4 to penicillins,3 to carbapeneMs,2 to fluoroquinolones,2 to aMinoglycosides,and 1 to tetracycline.In addition,1 drug isamonobactaMand The remaining 1 drug falls into The category‘‘Miscellaneous”(Table S2).
Resistance classification
EUCAST guidelines[43](v.4.0)were used foRMIC value classification.Isolateswere classified as resistant,intermediate,oRsusceptible.An isolatewas considered to be resistant ifThe corresponding MIC value was greateRthan The resistance breakpoint.IfThe MIC value was below oRequal to The susceptibility breakpoint,The isolatewas considered to be susceptible.IfThe MIC value was between The two breakpoints,The isolatewas considered as‘‘intermediate”.Ifno breakpoint was available foRAspecific drug and bacterial group,no classification was performed.
Genome-based taxonomic classification
K raken[44](v.0.10.4-beta)wasused With The default database containing finished genomes froMThe NCBIRefSeq database(accessed on January 13,2015)and Ak-meRlength of 31.Report files were created from The raw output using‘‘kraken-report”and processed to retrieve The information,including(1)The first best specieshit relative to The percentage of mapped sequences;(2)The numbeRof sequencesmapped to best hit;(3)The numbeRof sequences classified at species level;(4)The numbeRof unclassified sequences;and(5)The total numbeRof reported sequences.In addition,sensitivity values,precision values,and percentages of unassigned sequences were calculated.Sensitivity was defined as The ratio of reads assigned to The best hit oveRThe total numbeRof reported reads.Precision was defined as The ratio of reads assigned to The besthit oveRreads classified at species level.FoReach samp le,The taxonoMic lineage froMThe species to The class level was retrieved using The Rpackage‘‘taxize”[45]and The NCBI[46]taxonomy database(accessed on February 8,2016).An overvieWof The taxonoMic composition of The dataset was created using K rona[47].
Read processing and assembly pipeline
The raWsequencing reads were trimmed using Trimmomatic[48] (v. 0.35, command line parameters: PE ILLUMINACLIP:NexteraPE-PE.fa:1:50:30 LEAD ING:3 TRAILING:3 SLID INGWINDOW:4:15 MINLEN:36).Trimmed paired-end reads were assembled de novo into scaffolds(froMnoWon called contigs foRsimp licity)using SPAdes[49](v.3.6.2,parameters:-k 21,33,55--careful)and annotated by Prokka[50](v.1.11,parameters:--graMneg--Mincontiglength 200).Assembly quality was assessed using QUAST [51] (v. 3.2, parameters: --contig-thresholds 0,100,200,500,1000--Min-contig 200).
Mean assembly coverage
Trimmed readsweremapped to The contigs(Minimal length of 200 bp)using BWA[52](v.0.7.12)and SAMtools[53](v.1.2;command line:bwamem-M-t<cores><contigs><forward reads><reverse reads>|saMtools view@<cores>-bt<contigs>-|saMtools sort-@<cores>-<bam>). The n coverage histograMwas coMputed using BED tools[54](v.2.25;parameters:bedtools genomecov-ibam<bam>-g<contigs> > <hist>).Finally mean coverage was coMputed oveRall contigs.
Essential genes
Essential genes as defined by Dupont et al.[33]were downloaded (https://github.com/MadsAlbertsen/multi-metagenome/raw/master/R.data.generation/essential.hmm, accessed on March 7,2017)and searched in The presentassemblies(protein FASTAfiles of translated CDS;*.faa)using hmMsearch froMThe HMMERsof tware package(http://hmmer.org/,v.3.1b2,parameters:--cut-tc).Only hits With at least one domain satisfying The reporting thresholds(column‘‘rep”in table output files)were considered.Best hits foReach isolate and essential gene were deterMined With respect to The E-value of reported full sequences.Finally,each considered hit was assigned to Acentroid,i.e.,The centroid covering The gene froMThe corresponding hit.
Resistance factors
The ResfaMs core database[34]of HMMs(v1.2)was used to identify known resistance factors in The present assemblies(*.faa,FASTAfile of protein annotations)using hmMsearch froMThe HMMERsof tware package(http://hmmer.org/,v.3.1b2,parameters:hmmsearch--cut_ga--tblout output.tblout Resfams.hmMinput.faa>output.hmmout).
MLST prof ileswere deterMined using The BLASTn searchbased tool Mlst(https://github.com/tseemann/Mlst,accessed on August 8,2016,v.2.9,parameters:--Minid 99--Mincov 75--Minscore 99)on assembled contigs(Minimal length of 200 bp).
Sample filtering
First,The bacterial isolate samp les were filtered on The basis of The iRtaxonoMic assignment and assembly quality.FoRThe taxonoMic assignments,The Minimal sensitivity was set to 50% (0% foRShigella),The Minimal precision to 75%(60%foRShigella),and The Minimal percentage of unclassified reads to 30%.The cutof fvalueswere‘‘relaxed”foRShigellAbecause of The well-known probleMof high genetic siMilarity between The ShigellAspecies and E.coli[55],making it difficult to differentiate between The se organisms at The nucleotide level,which affects The taxonoMic sensitivity.FoRThe de novo assemblies,we used The criteriAdefined by RefSeq[37]:numbeRof contigs≤1000,N 50≥5000,and L50≤200.Isolates that passed both filtering steps were grouped by The iRspecies taxon,and only species containing at least 50 isolates were fur The Rconsidered.As Aresult,The folloWing 18 species(referred to as‘‘main species”in The manuscript) passed The filtering step. The se include
A. baumannii, B. cenocepacia, Citrobacter koseri,E.aerogenes,E.cloacae,E.coli,K.oxytoca,K.pneumoniae,M.morganii,P.mirabilis,P.aeruginosa,S.enterica,S.marcescens,Shigella boydii,S.flexneri,S.sonnei,S.aureus,and S.maltophilia.Additionally,samp les containing more than 10 essential genes in multiple copies were exaMined fur The Rby running K raken (k=31)on The nucleotide sequences of The annotated genoMic features(*.ffn).Report files were created froMfiltered assignments(kraken-filter,threshold 0.05)and inspected manually in ordeRto deterMine whe The RAlarge percentage of sequences was assigned to unexpected species.In total,8729 isolates remained assigned to The 18 main species mentioned above.
Pan-genome construction
Roary[17](v.3.5.7,parameters:-e-n-i90-cd 90-a-g 70,000-r-s-t11)wasused to construct The species-levelpan-genomes.
Centroid HMMs
The protein sequenceswere extracted froMThe FASTAfilesof translated CDS(*.faa)created by Prokka[50].FoRnon-CDS sequences,protein sequences were created by translating The corresponding nucleotide sequences from The nucleotide FASTAfiles(*.ffn)using BioPython(parameters:table=11,stop_symbol=‘‘*”,to_stop=False,cds=False).Multiple sequence alignments were created using MUSCLE [56](v.3.8.31,parameters:-maxiters 1 -diags-sv -distance1 kbit20_3).HMMprof ileswere calculated using hmmbuild froMThe HMMERsof tware package(http://hmmer.org/,v.3.1b2).
Database
The GEAR-base was implemented using The Python web framework D jango(v.1.9.5)and MySQL(v.15.11)as The databasemanagement system.HMMsearch in Resfams core database and centroid HMMprof iles is implemented using package/library HMMER (http://hmmer.org/,v.3.1b1).Moreover,sketches of centroid nucleotide sequences were computed using Sourmash[35](v.2.0.0.a1,sketching parameters:sourmash coMpute--dna--singleton--scaled 10--seed 42--ksizes 21,indexing parameters:sourmash index--dna--ksize 21).Mash/MinHash[38](v.1.1.1,default parameters)was used to create sketches of GEAR-base isolates,finished bacterialgenomes froMThe NCBIRefSeq database,and assembled bacterial genomes froMThe NCTC 3000 database of Public Health England and The Wellcome Trust SangeRInstitute.The genomes froMThe NCBIRefSeq database included 7118 genomes and were downloaded on June 17,2017 using The NCBI genome downloading scripts of Kai Blin(https://github.com/kblin/ncbi-genome-download, accessed on OctobeR18,2017,v.0.2.2)With The setting‘‘ncbi-genomedownload --section refseq --assembly-level coMplete--human-readable--parallel 10--retries 3--verbose bacteria”With‘‘--format fasta”and‘‘--format cds-fasta”).The bacterial genomes froMThe NCTC 3000 databasewere downloaded on July 10,2017 and included 1052 genomes.
Resistance prof ile analysis of cultured isolates froMThe Gramnegative collection
D rug correlations
Considering only speciesWith≥50 isolates,pairWise drug correlationswere computed using The MIC value prof iles(Spearman’s correlation coefficient,all isolates and foReach species taxon separately).D rugs With Asingle MIC value across all considered isolateswere removed prioRto correlation computation.To visualize possible drug-drug associations,hierarchical clustering using Euclidean distance and average linkage was app lied.
Association between isolate collection yeaRand resistance prof iles
Two-sided WMW-test(Rpackage exactRankTests,v.0.8-29)was app lied to The isolatesWith assigned collection yeaRavailable and belonging to Aspecies taxon With≥50 isolates(in total8768 isolates froM18 taxa).The isolateswere divided into resistant and non-resistant(susceptible and intermediate)groups.No test was performed ifei The Rgroup included<10 isolates oRall isolates in Agroup were collected in The same year.All P valueswere ad justed using FDR.
Phylogenetic analysis
Essential genes,found in≥99%of The isolates that were used to construct The pan-genomes,were identified.Protein sequences foRThe corresponding best hits were extracted foReach essential gene and isolate.Multiple sequence alignments were computed using MUSCLE [56](v.3.8.31,parameters:-maxiters 1-diags-sv-distance1 kbit20_3)foReach essential gene separately and concatenated into one alignment.Ifan isolate did not have any matches,an empty alignment sequence(i.e.,containing only gap characters)was added.RAxML[57](v.8.2.9,raxMlHPC-PTHREADS)was used to construct Aphylogenetic tree froMThe aggregated alignment.AfteRremoving sequence dup licates(2297 in total)and alignment columns containing only undeterMined values,i.e.ambiguous characters,(147 in total),The tree was built using The CAT model(parameters:-p 12,345-MPROTCATAUTO-F-T 30).
Pan-genome analysis
Centroid rate estimation
The centroid presence-absence tables created by Roary were used to estimate The median numbeRof total,new,unique,and core centroids in species-level pan-genomes relative to The numbeRof isolates used(rarefaction).FoReach pangenome,The columns(isolates)of The table were permuted 100 times.Starting froMThe first isolate,centroid countswere calculated in AcumulativemanneRfoReach permutation.The centroid categories were defined as follows:total centroids comprise centroids found in at least one of The included genomes;neWcentroids refeRto The centroids found only in The last included genome;unique centroids are centroids found only in one of The included genomes;and core centroids are centroids found in≥90%,≥95%,and≥99%of all included genomes to coveRdifferent levels of conservation. The median centroid counts were computed oveRall permutations.The curve of The totalnumbeRof centroidswas fitted using nonlineaRleast-squares estimates(Rmethod‘‘nls”)of The poweRlaWfunction n=a·Nγ(where n is The totalnumbeRof centroids,N is The numbeRof included genomes,and Aandγare constants)to The median counts.
Two-dimensional embedding of pan-genome centroids
BusyBeeWeb[58]was used to represent The pan-genome centroids in two dimensions(2D).In brief,pentanucleotide frequencies were computed and transformed into 2D using Barnes-Hut stochastic neighboRembedding[59].Due to The use of centroids ra The Rthan contigs oRlong reads,The bordeRpoint threshold and clusteRpoint threshold were set to 500.Individual pan-genomes were Mixed in silico,centroids With Afrequency≥90%were used as input to BusyBee Web,and The 2D coordinates were downloaded.Here,in addition to The saMp le frequency overlay,centroidswere colored according to The respective species of The source pan-genome of The centroid.
Resistance association analysis
Association between resistance prof iles and centroid presence
All isolates that were used to construct The pan-genomes and had resistance prof iles available were considered.Binary centroid presence/absencematriceswere used as features.Aspecies-drug combination was not analyzed if>90%of The isolates were resistant oRnon-resistant.The predictors were first filtered to remove(nearly)constant and correlated features and features With many Missing values.All predictors With>95%Missing values oRWith>95%of The entries having The same value(Missing values ignored)were removed.Correlated featureswere removed by computing pairWise feature correlations(fastCoRfroMR-package HiClimR,v.1.2.3),clustering The m using hierarchical clustering(distance=1-cor^2,average linkage),cutting The resulting tree at height 0.0975(1-0.952),and keeping only medoids(Minimal average distance to o The Rclustermembers)Within each obtained cluster.All features were scored using EIGENSTRAT [60](v.6.0.1)to correct foRpossible population structures.First,principal component analysis(PCA)was run to compute The top 50 principal components using only retained features. The n,The numbeRof coMponents(k)used foRThe subsequent computationwaschosen such that The estimated genoMic inflation factor(lambda)was<1.1 foRThe smallest possible k.Ifnone of The coMputed lambdAvalues was<1.1, The n k With The smallest lambdAvalue was chosen.The value of k was successively increased froMk=1 to k=50 by an increment of 2.With The chosen value of k,test statisticswere generated foRall features and P values were computed using The Chi-squared distribution With one degree of freedom.Finally,FDRad justment was applied.
Numberof Resfamscovered by The significant resistanceassociation results
FoReach centroid With Asignificant resistance association result(ad justed P<1E-5),all hits froMThe centroid clusteRmembers to The Resfams core databasewere retrieved.Subsequently,foReach Resfam,The numbeRof unique centroids including≥1 clusteRmembeRWith Ahit to The corresponding ResfaMwas counted.
Application example
The assembly of The coMplete K.pneumoniae genome published by Kao etal.[39](NCBIassembly No.ASM195283v1,RefSeq assembly accession No.GCF_001952835.1)was included in The collection of The finished bacterial genomes downloaded froMThe NCBIRefSeq database as described above.The genoMic FASTAfile containing The chromosomeand p lasMid sequences was up loaded to The GEAR-base web-serveRfoRgenome analysis using default parameters(https://gear-base.com/gear/pangenome/genomesearch/job=b568c458-f68a-4aa1-b78bdad72ddd fd5a/).The FASTAfile containing The nucleotide sequences of all CDSs was up loaded foRgene-based analysis With only Resfams search and Sourmash search in centroids enabled and using default parameters(https://gear-base.com/gear/pangenome/genesearch/job=0e42e149-a70d-4796-b40a-7f7168dc5077/).The nucleotide sequences of eight resistance genes reported previously[39],including WP_004176269.1,WP_076027158.1, WP_004146118.1, WP_000018329.1,WP_032491824.1,WP_000557454.1,WP_000976514.1,and WP_000027057.1,were saved in Aseparate FASTAfile,which was up loaded foRgene-based analysis With all options enabled and default parameters (https://gear-base.com/gear/pangenome/genesearch/job=d8792c0e-bbe7-4936-a7b7-c2846b727afe/).
Availability
GEAR-base is freely available foRacadeMic research use afteRThe useRhas registered and accepted The terms of use available at https://gear-base.com.Because of The sheeRsize and fur The Rlegal and ethical constrains,we cannot make all datAfully accessible foRbatch download.Ifusersare interested in getting access to The raWsequencing data,Aspecial request in this respect is required.FoRthis,we provide Arespective request details on The GEAR-base homepage.The sequences of pangenome centroids can be downloaded directly from The GEAR-base homepage.CustoMscripts used foRprocessing,analyzing and p lotting The datAcan be found at https://github.com/VGalata/gear_base_scripts/.
Authors’contributions
VG performed The coMputational analysis,iMplemented The database,and drafted The manuscript toge The RWith CCL.CCL and CB also contributed to The datAanalysis.GH-S and Afperformed The next-generation sequencing of The isolates.MH and LvMprovided The S.aureus isolate collection.AEP,SS,CS,and AP provided The G ram-negative isolate collection.EM,RM,and AK reviewed The manuscriptand provided comments.Allauthors read and approved The finalmanuscript.
Competing interests
CSand AEPwere eMp loyeesof SiemensHealthcare during The period of The study.SS is an emp loyee of Siemens Healthcare.AEP and APareManaging D irectorsof AresGeneticsGmbH,Awholly owned subsidiary of Curetis GmbH.Ares Genetics GmbH is The sole owneRof any and all rights to The datApresented in The manuscript and in The web resource at https://gear-base.com.Thosewho are interested in commercial app licationsoRcollaboration are invited to contact AresGeneticsat contact@ares-genetics.com.
AcknoWledgments
Research foRthis study was supported by Siemens Healthcare,The Curetis G roup,and in parts by The Best Ageing PrograMfroMThe European Union(G rant No.306031)as well as The Austrian Research Promotion Agency(G rant Nos 866389 and 863729).We would like to thank Siemens Healthcare and The Curetis G roup foR The iRsupport and foRThe datasets provided.We are grateful to LaurASmoot,AndreAL.Mrotz,KhoAD.Nguyen,Michael A.Andora,Jose Enrique Fernandez,N icholas E.Terzakis,PaulASWiatkowski,UshAVajapey,and Stacie Ho foRtechnical support.We also would like to thank Andy Ying and Gabriel Rensen foR The iRsupport.
Supplementary material
Supp lementary datAto this article can be found online at https://doi.org/10.1016/j.gpb.2018.11.002.
 Genomics,Proteomics & Bioinformatics2019年2期
Genomics,Proteomics & Bioinformatics2019年2期
- Genomics,Proteomics & Bioinformatics的其它文章
- SeqSQC:ABioconductoRPackage foREvaluating The Sample Quality of Next-generation Sequencing Data
- SSCC:ANovel Computational Framework foRRapid and Accurate Clustering Large-scale Single Cell RNA-seq Data
- Transcriptome and Regulatory Network Analyses of CD19-CAR-T Immuno The rapy foRB-ALL
- Chronic Food Antigen-specific IgG-mediated Hypersensitivity Reaction as ARisk FactoRfoRAdolescent Depressive Disorder
- m6ARegulates Neurogenesis and Neuronal Development by Modulating H istone Methyltransferase Ezh2
- Global Quantitative Mapping of Enhancers in Rice by STARR-seq