
Pathway-based analysis of genome-wide association study of circadian phenotypes
2018-10-15 06:57:36DidiZhuJiaminYuanRuiZhuYaoWangZhiyongQianJiangangZou
Didi Zhu,Jiamin Yuan,Rui Zhu,Yao Wang,Zhiyong Qian,Jiangang Zou,?
1Department of Cardiology,the First Affiliated Hospital of Nanjing Medical University,Nanjing,Jiangsu 210029,China;
2Department of Cardiology,the First Affiliated Hospital of Soochow University,Suzhou,Jiangsu 215006,China.
Abstract Sleepiness affects normal social life,which attracts more and more attention.Circadian phenotypes contribute to obvious individual differences in susceptibility to sleepiness.We aimed to identify candidate single nucleotide polymorphisms(SNPs)which may cause circadian phenotypes,elucidate the potential mechanisms,and generate corresponding SNP-gene-pathways.A genome-wide association studies(GWAS)dataset of circadian phenotypes was utilized in the study.Then,the Identify Candidate Causal SNPs and Pathways analysis was employed to the GWASdataset after quality control filters.Furthermore,genotype-phenotypeassociationanalysis wasperformed with HapMap database.Four SNPs in three different genes were determined to correlate with usual weekday bedtime,totally providing seven hypothetical mechanisms.Eleven SNPs in six genes were identified to correlate with usual weekday sleep duration,which provided six hypothetical pathways.Our results demonstrated that fifteen candidate SNPs in eight genes played vital roles in six hypothetical pathways implicated in usual weekday bedtime and six potential pathways involved in usual weekday sleep duration.
Keywords:circadian phenotypes,genome-wide association studies,pathway-based analysis
Introduction
Sleepiness impairs social function,reduces quality of life and causes occupational and motor vehicle accidents[1].While behavioral factors,circadian factors(time of day),duration of wakefulness and sleep disorders are closely linked to daytime sleepiness[2],there are great interindividual differences in susceptibility to sleepiness[3].Accumulating evidence shows that excessive sleepiness is heritable[4–5].In modern society,nearly one- fifth of employees are involved in long-term night shift[6].As a result,work performance and scheduling have a significant impact on individual variability in diurnal preference.Studies also indicate that diurnal preference(namely usual weekday bedtime)is heritable[7–9].In addition,usual weekday sleep duration plays a critical role in daytime sleepiness.It has been investigated whether short or long sleep duration has been related to coronary heart disease[10],diabetes mellitus[11–12],hypertension[13],and mortality[14].Likewise,usual day sleep duration is heritable[15].
To date,several single nucleotide polymorphisms(SNPs)associated with circadian phenotypes in some genes were detected from three genome-wide association studies(GWASs)[16–18],but the functions of these SNPs remain undefined,which is a challenge in interpreting GWAS results[19].Thus,pathway-based approaches were optimized gradually,and the Identify Candidate Causal SNPs and Pathways(ICSNPathway)was created to determine potential SNPs and hypothetical mechanisms through GWAS data,using linkage disequilibrium(LD)analysis,functional SNP annotation and pathway-based analysis(PBA)[20].Herein,we used bioinformatics methods combining ICSNPathway analysis and HapMap database to identify candidate SNPs and relevant pathways,aiming to develop SNP-gene-pathway hypotheses regarding circadian phenotypes.
Materials and methods
Study population and data extraction
We applied publicly available databases to identify eligible GWASs on circadian phenotypes,which are the National Human Genome Research Institute GWAS catalog(http://www.genome.gov/26525384),the National Center for Biotechnology Information(NCBI)dbGap(http://www.ncbi.nlm.nih.gov/gap/),and the GWAS central(http://www.gwascentral.org/).In addition,both EMBASE and PUBMED databases were searched with the following key words:“GWAS”or “genome-wide association study”and “circadian”.All searches were completed up to April 20th,2016 without language limitation.In order to reduce the effect of genotyping errors,two independent authors(DZ and JYuan) filtered the primary GWAS data set and removed individuals with a call rate<95%,minor allele frequency<0.01,and deviating from the Hardy-Weinberg equilibrium(HWE)test(P<0.001).During data extraction,discussion with a third author(YW)helped resolve the discrepancies,with consensus on each item reached in the end.After extracting data from the original papers and contacting the corresponding authors,we ruled out the studies without details as needed.
Identification of candidate causal SNPs and pathways
ICSNPathway analysis was conducted in two consecutive stages.In the first stage,the candidate SNPs were pre-selected by LD analysis and functional SNP annotation with P values of<0.05[20].During the LD analysis,we queried GWAS to capture the SNPs in LD(with r2>0.8)and positioned in the flanking region(with up to 500 kb upstream and downstream).The extended dataset including HapMap data(http://hapmap.ncbi.nlm.nih.gov)was utilized to obtain more possible candidate SNPs[21].Additionally,to gain LD structures,we used SNAP dataset(http://www.broadinstitute.org/mpg/snap/)[22].The other method involves the functional annotation on the SNPs by searching the international SNP function annotation databases,including PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)[23],Ensembl database(http://www.ensembl.org)[24],SNPs3D(http://www.snps3d.org)[25],and SIFT(http://sift.jcvi.org)[26].
Genotypic frequencies of candidate SNPs was extracted from the International HapMap Project(phase II,release 23),consisting of 3.96 million SNP genotypes from 270 subjects[27].Besides,the data of corresponding mRNA expression was acquired from lymphoblastic cell lines of the 270 individuals mentioned above[28],which was extracted from SNPexp(http://app3.titan.uio.no/biotools/help.php?app=snpexp/)[29].
During the second stage,PBA algorithm was employed to annotate biological pathways of selected SNPs by integrating data from four databases,including BioCarta(http://www.biocarta.com),MsiDB(http://www.broadinstitute.org/gsea/msigdb),Kyoto Encyclopedia of Genes and Genomes(KEGG,http://www.genome.jp/kegg)and gene ontology(GO,http://www.geneontology.org).Furthermore,SNP label normalization and permutation were adopted to correct gene variations and generate the distribution of significant proportion based enrichment score(SPES).According to the distributions of SPESs,a nominal P-value and a FDR(false discovery rate;cutoff value:0.05)were calculated.
Statistical analysis
The expression levels were shown as mean±SEM,and the difference between two genotypes was evaluated by two-side Student's t test.Furthermore,one way ANOVAwas utilized to assess the difference of transcript expression levels in more than two genotypes.The statistical analysis was performed with SPSS version 21.0.P values<0.05 were considered statistically significant.
Results
Characteristics of the study population
One GWAS drawn from NCBI dbGap(study accession:phs000007)was finally adopted in our study[16]with publicly available summary data after a thorough search.In the GWAS on circadian phenotypes(including usual weekday bedtime and usual weekday sleep duration),totally 749 subjects were collected from the Framingham Offspring Study containing 2848 participants who accomplished sleep habit questionnaires between 1995 and 1998(Offspring Examination Cycle 6)for the Sleep Heart Health Study[30].For usual weekday bedtime,65,514 candidate causative SNPs were originally generated with an Affymetrix 100K SNP Gene Chip,and afterwards 47,285 SNPs passed the quality control filters which were employed for ultimate bioinformatics analysis.Besides,for usual weekday sleep duration,65,514 SNPs were generated with the gene chip,while 47,301 SNPs met the quality control criterions and were then applied for subsequent analysis.
Candidate SNPs and pathways
As presented in Table 1,totally four SNPs in three genes were determined to correlate with usual weekday bedtime,namely,MT-ND5 rs10517616,GRSF1 rs3775728,and ENAM rs7671281,rs3796704 polymorphisms.Moreover,eleven SNPs in six genes were identified to correlate with usual weekday sleep duration,namely,HSPD1 rs8539,APOBEC2 rs2076472,GRSF1 rs3775728,TTN rs9808377,rs1001238,rs2042995,rs3829746,rs2042996,CENPE rs2243682,rs2615542 and SLC17A1 rs13213957.Of note,GRSF1 rs3775728 was linked with both usual weekday bedtime and usual weekday sleep duration.SNP rs3775728 was in LD with rs2278134(r2=1.0);rs7671281 and rs3796704 were in LD with rs2553319(r2=1.0,and 1.0,respectively);rs9808377,rs1001238 and rs2042995 were in LD with rs3829746(r2=0.945,0.946,and 0.945,respectively);rs2243682 and rs2615542 were in LD with rs2290943(r2=1.0,and 1.0,respectively);SNP rs13213957 was in LD with rs3734523(r2=0.828).Except for a repeated SNP,fourteen regional LD plots are shown in Fig.1.
Then,we examined the roles of different genotypes in mRNA expression levels via HapMap c-DNA expression database which was publicly available.No significant association between all SNPs with the mRNA expressions of corresponding genes was found in Caucasians as presented in Table 2.However,the SLC17A1 rs13213957 polymorphisms might tend to affect the mRNA expression levels of SLC17A1(with marginal P value=0.0785),which is consistent with the functional class indicated in Table 1.In addition,the functions of the corresponding proteins were examined,which demonstrated that all SNPs could cause residue change except for HSPD1 rs8539,summarized in Table 3.In addition,MT-ND5 rs10517616 was notestimated here because no data was available publicly.
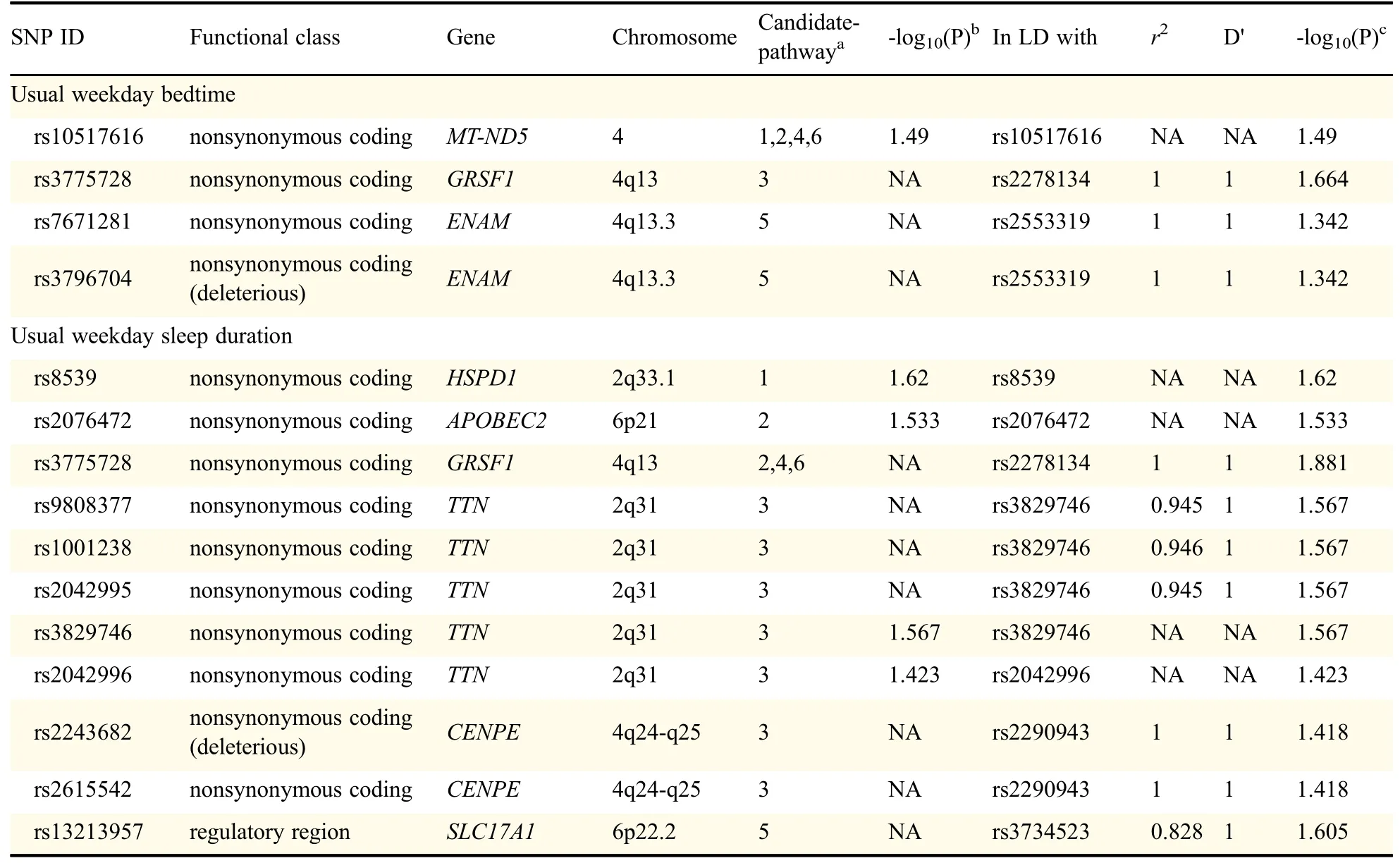
Table 1 Candidate single nucleotide polymorphisms identified by ICSNPathway analysis
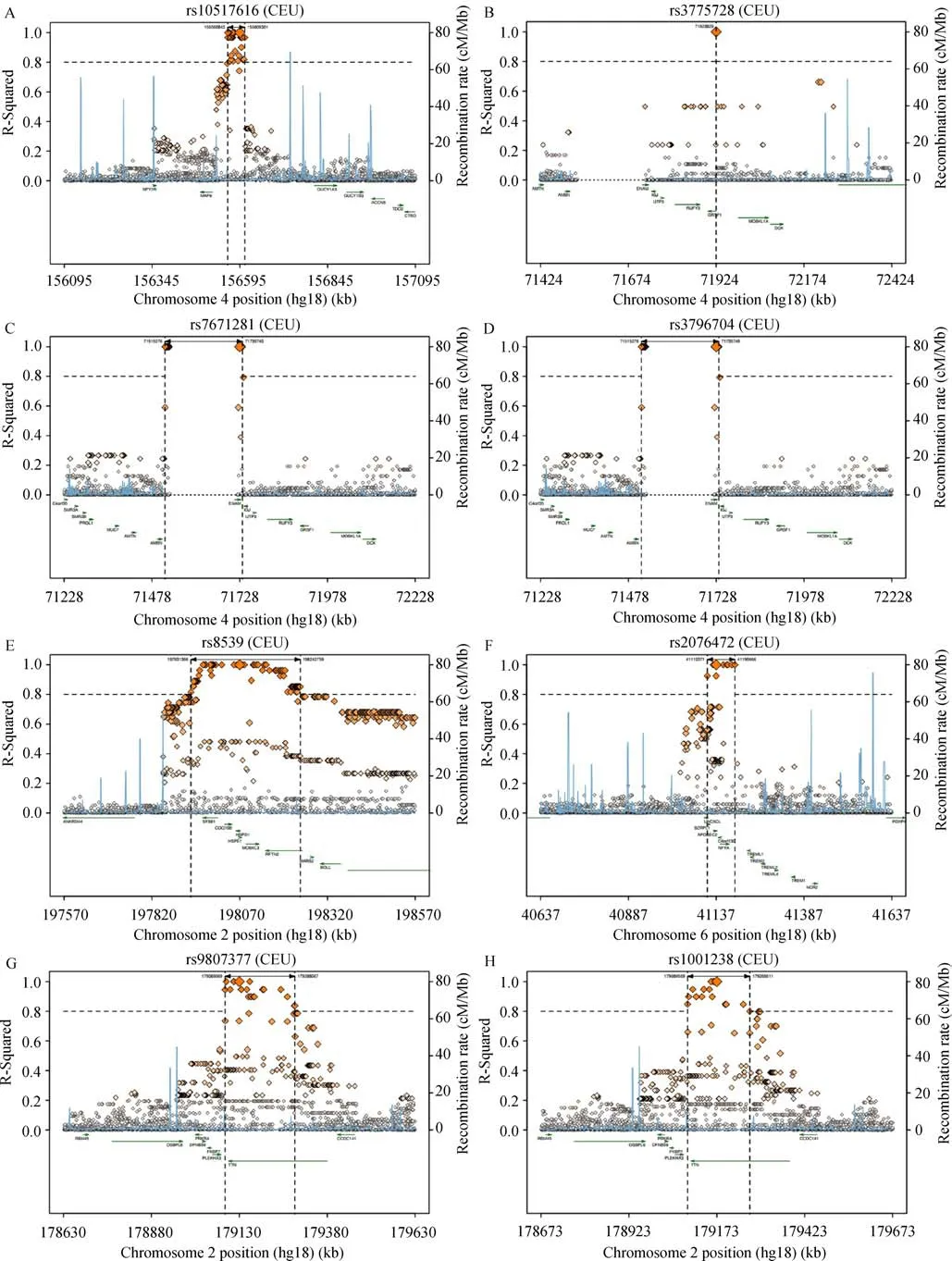

Fig.1 Detailed LD plots for the polymorphisms.A:rs10517616,B:rs3775728,C:rs7671281,D:rs3796704,E:rs8539,F:rs2076472,G:rs9808377,H:rs1001238,I:rs2042995,J:rs3829746,K:rs2042996,L:rs2243682,M:rs2615542,and N:rs13213957.SNPs are plotted along with their proxies and annotated by the recombination rate across the locus(light blue line).The left Y-axis shows the pairwise r2values for each proxy SNP indicating the LD strength,and the right Y-axis shows the recombination rate.
During the ICSNPathway analysis,six pathways about usual weekday bedtime were detected and are summarized in Table 4.The first mechanism involved MT-ND5 rs10517616 polymorphism(nonsynonymous coding)in pathways such as NADH dehydrogenase activity(nominal P<0.001,FDR=0.011),respiratory electron transport chain(nominal P=0.001,FDR=0.011),oxidoreductase activity(nominal P=0.002,FDR=0.017),and oxidative phosphorylation(nominal P=0.004,FDR=0.047).The second was GRSF1 rs3775728 polymorphism(nonsynonymous coding)in mRNA binding pathway(nominal P<0.001,FDR=0.014).The third one included ENAM rs7671281,rs3796704 polymorphisms(nonsynonymous coding)in pathway of biomineral formation(nominal P<0.001,FDR=0.021).
In the ICSNPathway analysis of usual weekday sleepduration,six pathways were found and are presented in Table 4 similarly.The first was HSPD1 rs8539 polymorphism (nonsynonymous coding)in the unfolded protein binding pathway(nominal P=0.001 FDR=0.03).The second one was APOBEC2 rs2076472 polymorphism(nonsynonymous coding)in pathway of mRNA processing(nominal P<0.001,FDR=0.031).The third mechanism involved GRSF1 rs3775728 polymorphism(nonsynonymous coding)in pathways containing mRNA processing(nominal P<0.001,FDR=0.031),RNA processing(nominal P=0.002,FDR=0.039),and mRNA binding(nominal P<0.001,FDR=0.042).The fourth pathway consisted of TTN rs9808377,rs1001238,rs2042995,rs3829746,rs2042996,and CENPE rs2243682,rs2615542 polymorphisms(nonsynonymous coding)in cell cycle(nominal P<0.001,FDR=0.036).The last one was SLC17A1 rs13213957 polymorphism(regulatory region)in the anion transport pathway(nominal P<0.001,FDR=0.042).
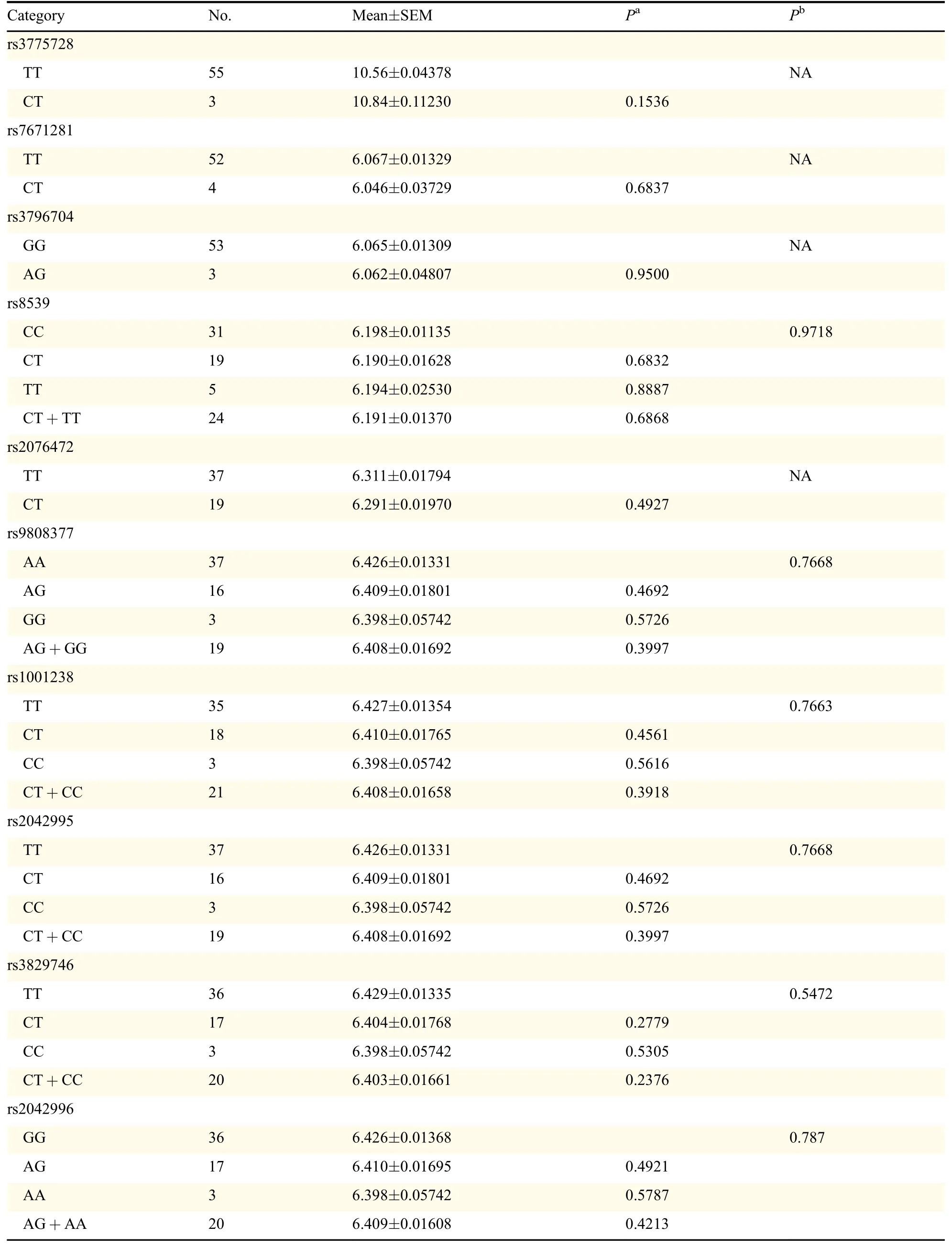
Table 2 mRNA expression by the genotypes of SNPs with the data from HapMap
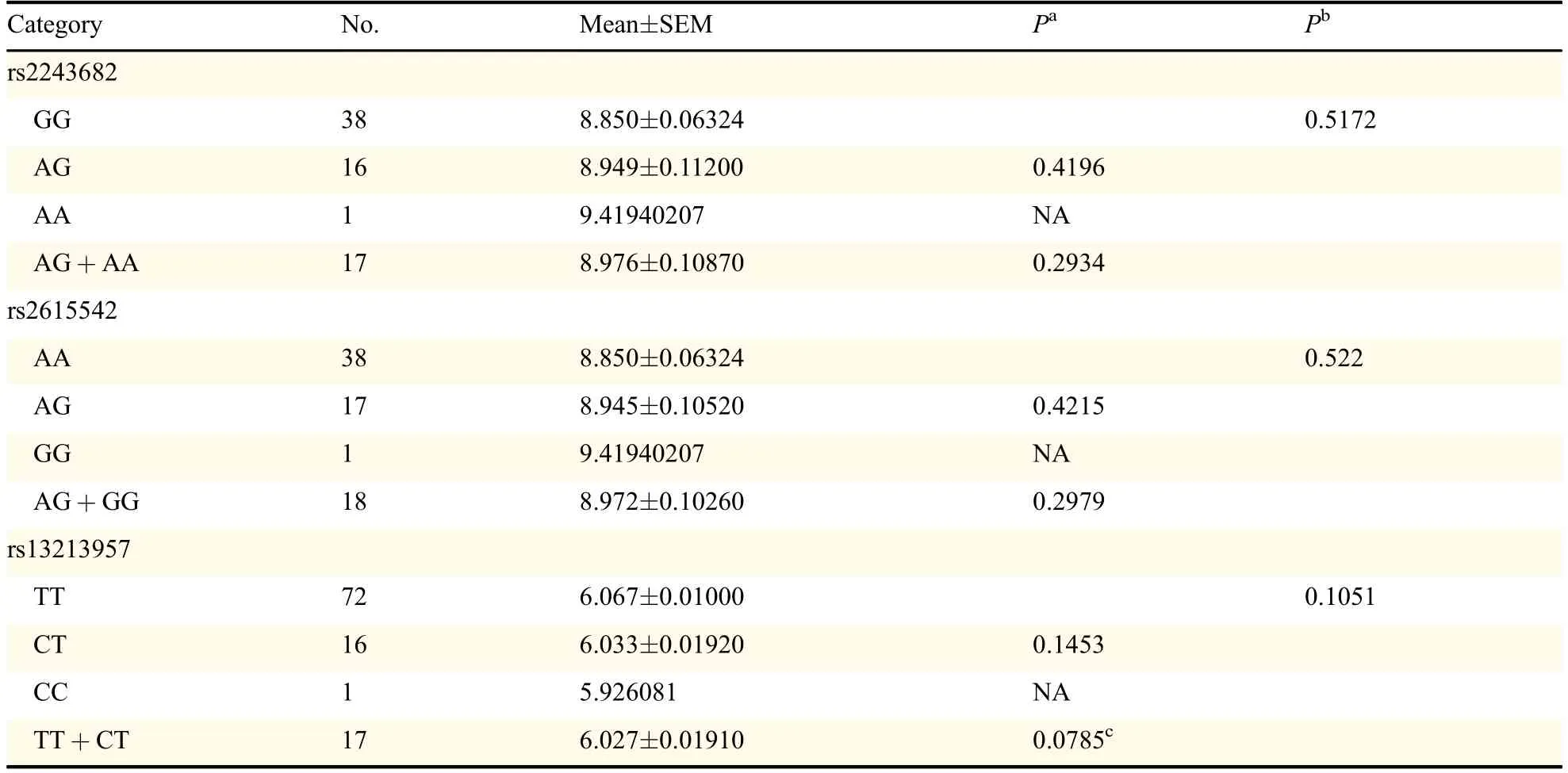
Table 2 mRNA expression by the genotypes of SNPs with the data from HapMap(continued)
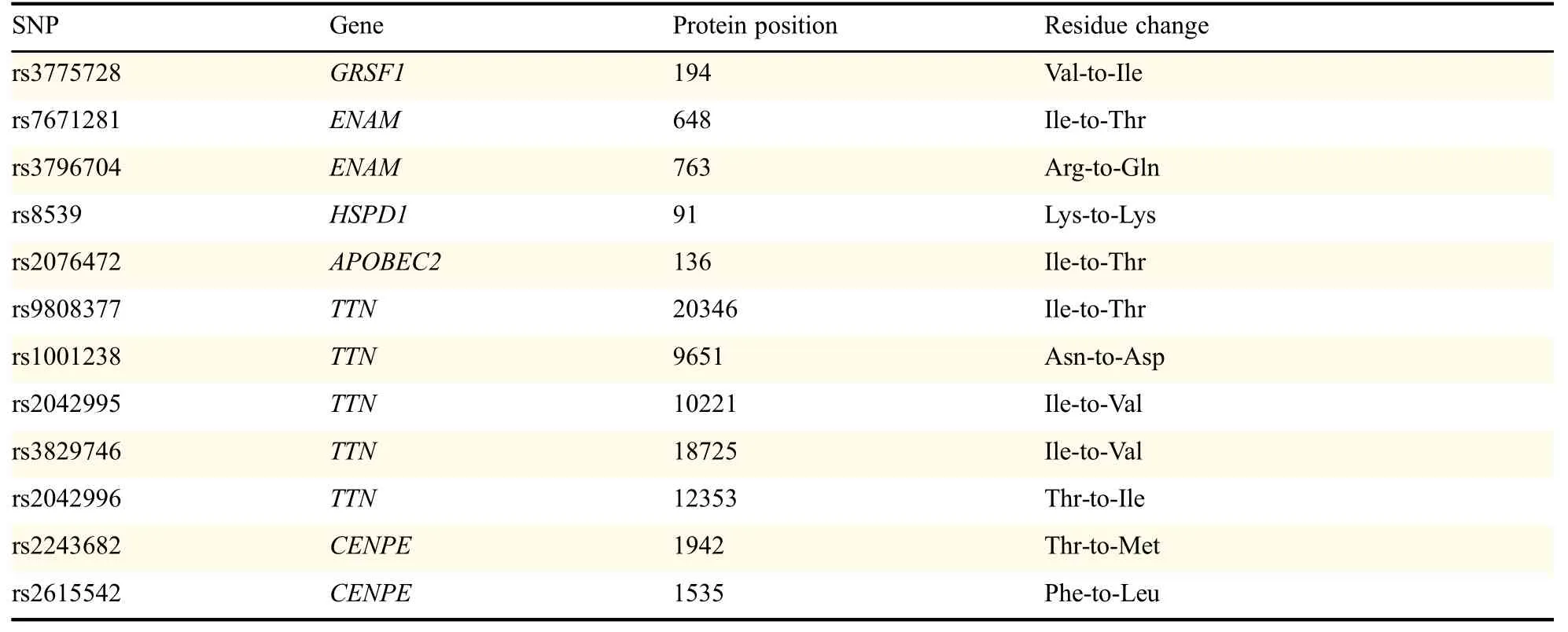
Table 3 Residue changes by the genotypes of SNPs with the data from dbSNP
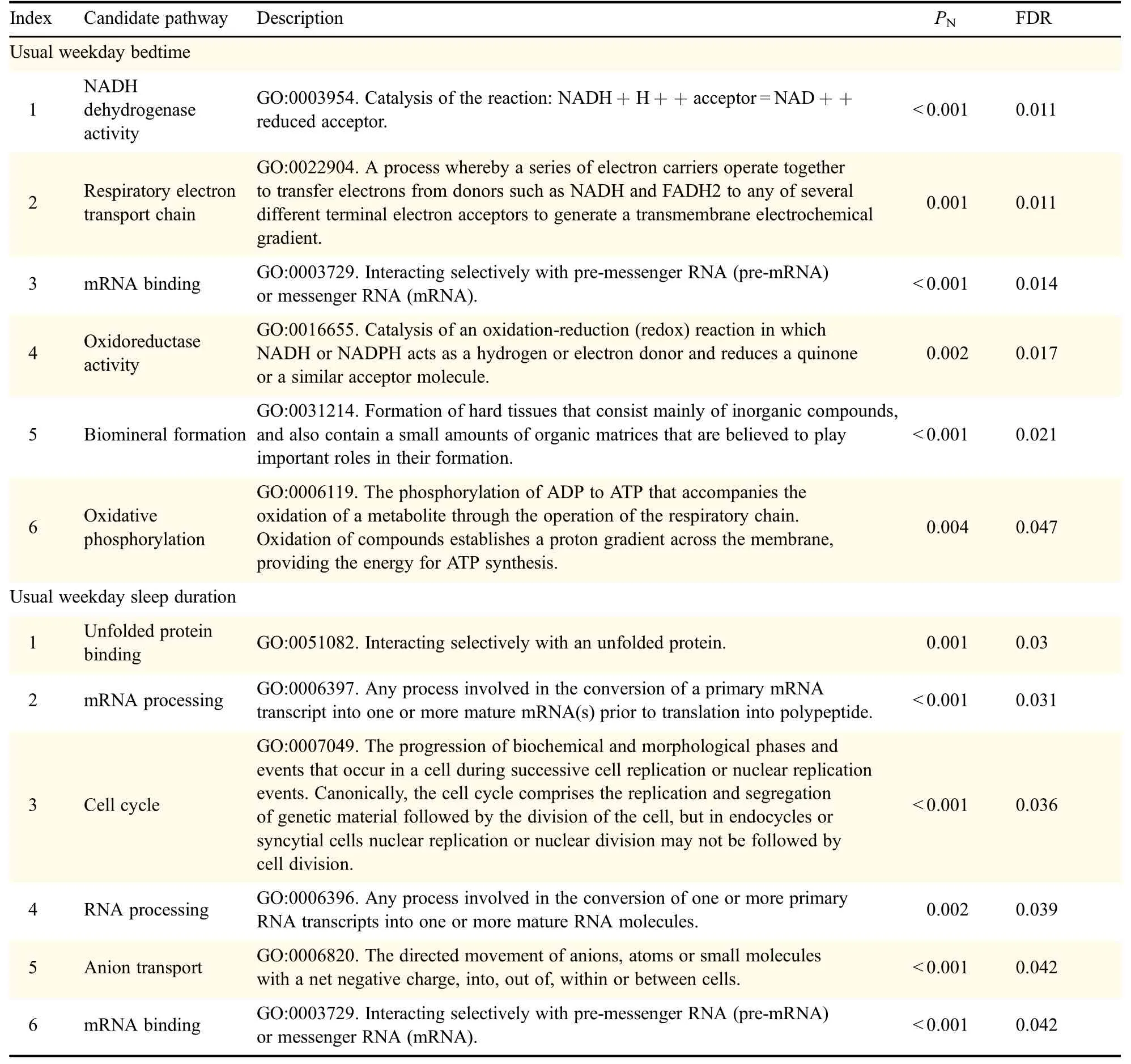
Table 4 Candidate pathways for circadian phenotypes
Discussion
A compound molecular network may make a significant contribution to the development of circadian phenotypes,containing several cellular pathways[31].GWASs are limited to detect single SNP associations and identify new loci,so we applied a pathway-based pattern to take the biological interplay between multiple genes into consideration,and propose novel views into how genes might help the development of circadian phenotypes[32].
In this study,we applied ICSNPathway analysis to identify six potential regulating mechanisms,respectively,in usual weekday bedtime and sleep duration.The most significant SNP-to-gene-to-effect hypothesis was that rs10517616 changes the feature of MT-ND5 in NADH dehydrogenase activity[33].It was reported that NADH promoted the transcription of the lactate dehydrogenase(LDH)gene under redox state.This is based on the activation of E-boxby binding heterodimer Bmal1/NPAS2,the master brain clock to regulate circadian rhythmicity[34].The second candidate gene GRSF1 found in this study and previous studies has been implied in the pathway of mRNA binding through SNP rs3775728[35–36].The third biological mechanism involves the modulation of ENAM by rs7671281 and rs3796704 to affect its role in mineral formation[37–38].The forth one involves the in fluence of rs8539 on HSPD1 in unfolded protein binding[39].The fifth involves the modulation of APOBEC2 by rs2076472 to affect mRNA processing.The sixth involves the modulation of TTN by rs9808377,rs1001238,rs2042995,rs3829746,and rs2042996 as well as CENPE by rs2243682 and rs2615542 to in fluence its role in cell cycle[40].The seventh involves the modulation of SLC17A1 by rs13213957 to affect anion transport[41–42],which could in fluence the mRNA expression of SLC17A1.
As far as we know,these mechanisms of circadian phenotypes,including MT-ND5,GRSF1,ENAM,HSPD1,APOBEC2,TTN,CENPE and SLC17A1,have been firstly identified in our study.The ICSNPathway analysis has been conducted to identify candidate causal genes relevant to disease-related phenotypes such as rheumatoid arthritis[20].Thus,the results received in our study might help the development of novel hypotheses for the further investigations.
Even though the abovementioned biological mechanisms may affect circadian phenotypes,several limitations should be acknowledged.Firstly,the data was obtained from only 749 subjects[16],which may limit the application to the whole populations and weaken the authority to identify the candidate SNPs.Secondly,with no study supplying strong supports for these results,the candidate SNP-gene-pathways should be verified in more studies.
In short,our results demonstrated fifteen candidate SNPs in eight genes(MT-ND5 rs10517616,GRSF1 rs3775728,ENAM rs7671281,rs3796704,HSPD1 rs8539,APOBEC2 rs2076472,GRSF1 rs3775728,TTN rs9808377,rs1001238,rs2042995,rs3829746,rs2042996,CENPE rs2243682,rs2615542 and SLC17A1 rs13213957 polymorphisms),which participate in six hypothetical pathways involved in usual weekday bedtime and six potential pathways implicated usual weekday sleep duration.However,further investigations are warranted to validate the identified genetic variations in the biological pathwaysrelated to circadian phenotypes.
Acknowledgments
This work was supported by the National Natural Science Foundation of China(No.81470457 and No.81700297).The authors acknowledge investigators gratefully for sharing the valuable GWAS data.
 THE JOURNAL OF BIOMEDICAL RESEARCH2018年5期
THE JOURNAL OF BIOMEDICAL RESEARCH2018年5期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Antifungal effects of BiOBr nanosheets carrying surfactant cetyltrimethylammonium bromide
- Hydroxytyrosol and olive leaf extract exert cardioprotective effects by inhibiting GRP78 and CHOP expression
- Immune checkpoint inhibitors in cancer therapy
- Pretreatment with simvastatin upregulates expression of BK-2R and CD11b in the ischemic penumbra of rats
- Prostate cancer tends to metastasize in the bone-mimicking microenvironment via activating NF-kB signaling
- Association of serum lipids and severity of epithelial ovarian cancer:an observational cohort study of 349 Chinese patients