
Animal models for filovirus infections
2018-03-24 04:35:44VinayakumarSiragamGaryWongXiangGuoQiu
Zoological Research 2018年1期
Vinayakumar Siragam, Gary Wong,3,4, Xiang-Guo Qiu,*
1 Special Pathogens Program, Public Health Agency of Canada, Winnipeg, Manitoba R3E 3R2, Canada
2 Department of Medical Microbiology, University of Manitoba, Winnipeg, Manitoba R3E 0J9, Canada
3 Shenzhen Key Laboratory of Pathogen and Immunity, State Key Discipline of Infectious Disease, Shenzhen Third People’s Hospital,Shenzhen Guangzhou 518020, China
4 Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101,China
lNTRODUCTlON
The family Filoviridae consists of three genera, Ebolavirus,Marburgvirus, and Cuevavirus (Kuhn et al., 2011, 2013).Filoviruses are classified as biosafety level 4 (BSL-4)pathogens and infections result in severe hemorrhagic fevers in humans and non-human primates (NHPs), with fatality rates as high as 90% (Sanchez et al., 2007). Marburgvirus has two member viruses: Marburg virus (MARV) and Ravn virus (RAVV).Ebolavirus has five member viruses: Ebola virus (EBOV),Sudan virus (SUDV), Tai Forest virus (TAFV), Bundibugyo virus(BDBV), and Reston virus (RESTV) (Kuhn et al., 2013). Live viruses belonging to Cuevavirus have not yet been isolated.Currently, no approved vaccines or therapeutics are available against filovirus infections; however, clinical trials of candidate vaccines and therapeutics conducted during the 2014–2016 EBOV outbreak in West Africa (Martin et al., 2016; Wong &Kobinger, 2015) provide hope that a licensed medical countermeasure is on the horizon.
The transmission of filoviruses to humans is poorly understood, but outbreaks are likely started by direct contact with dead animal carcasses (typically NHPs) or in the case of MARV, the bat reservoir, resulting in subsequent transmission within the susceptible human population (Leroy, 2009). The blood and/or other body fluids of dead animals and humans are especially infectious, with viremia in human patients reaching 108copies/mL of blood (Faye et al., 2015). Contact with blood and bodily fluids is thought to be the main mode of transmission. After an incubation period of 2–21 days, general symptoms such as fever, chills, fatigue, headache, and myalgia appear. As disease progresses, systemic (prostration,lethargy), gastrointestinal (anorexia, vomiting, abdominal pain,diarrhea), respiratory (chest pain, breath shortness, cough,nasal discharge), vascular (conjunctival injection, postural hypotension, edema), and neurological (headache, confusion,coma) symptoms may appear, sometimes accompanied with hemorrhage from venipuncture sites. In fatal cases, patients succumb after multiple organ failure between 6–16 days after the onset of symptoms (Nakayama & Saijo, 2013).
Animal models against filoviruses have been developed in mice, guinea pigs, hamsters, ferrets and NHPs (Bente et al.,2009; Bradfute et al., 2012; Connolly et al., 1999; Marzi et al.,2016; Kozak et al., 2016; Kroeker et al., 2017b; Yamaoka et al.,2017). The development of animal models that recapitulates hallmarks of human filoviral disease is crucial in understanding the pathogenesis of these viruses. In this review, we summarize the animal models that have been developed for filoviruses(Table 1), and discuss the advantages and disadvantages of each animal model (Table 2).

Table 1 Animal models for studying filovirus infections

Table 2 Advantages and disadvantages of animal models for filovirus infections
MOUSE MODELS
Mice are easy to handle in the laboratory, are available commercially in large numbers, and have a low unit cost. Due to the wide availability of biochemical reagents and immunological tools, mice are preferred as the small animal model for filovirus research (Bradfute et al., 2007).
lmmunocompetent Mouse Model
Ebola virus (EBOV)
As immunocompetent mice are resistant to infections with wild type EBOV (WT-EBOV) (Banadyga et al., 2016; Bray, 2001;Warfield et al., 2009), Bray et al. (1998) adapted the 1976 Mayinga isolate of EBOV to cause lethal disease in adult immunocompetent mice through the sequential passaging of infected liver homogenates in newborn mice. The resultant plaque-purified mouse-adapted EBOV (MA-EBOV) was lethal for adult BALB/c, CD-1, and C57BL/6 mice at 30 or 3 000 median lethal dose (LD50) of 100or 102plaque-forming units(pfu) when injected intraperitoneally (i.p.). Remarkably, infection with MA-EBOV did not produce any disease symptoms via the subcutaneous (s.c.) or intramuscular (i.m.) routes at doses of 106pfu (Bray et al., 1998). Compared with WT-EBOV, MAEBOV exhibited eight amino acid changes in the coding and non-coding regions of the virus genome (Ebihara et al., 2006),with amino acid changes observed in viral protein (VP) 35,VP24, nucleoprotein (NP), and RNA-dependent RNA polymerase(L) genes. Furthermore, the determinants of virulence in mice were shown to be mutations in NP and VP24, in which recombinant viruses containing only the NP and VP24 mutations were resistant to type I interferon (IFN) in vitro and conferred lethality to mice (Ebihara et al., 2006). The disease observed in adult immunocompetent mice by i.p. inoculation of MA-EBOV closely reflected EBOV infection in humans, and the virus replicated well in blood, reaching peak titers of109pfu/mL.Biochemical and histopathological studies have shown diminished functions in the liver and kidney (Gibb et al., 2001b).In addition, lymphocyte apoptosis and decrease in platelet counts were observed in mice infected with MA-EBOV. In livers of MA-EBOV-infected mice, viral replication was seen in hepatocytes, Kupffer cells, and sinusoidal endothelial lining cells. In the spleen, viral antigen was noted at day 2 after infection. Coagulopathy, such as disseminated intravascular coagulation with prolongation of prothrombin time (PT) and activated partial thromboplastin time (APTT) have also been detected at advanced stages of the disease (Bray et al., 2001;Warfield et al., 2009). However, fibrin deposition and breakdown at sites of viral replication in the spleen and other tissues have not been observed in tissue sections of MAEBOV-infected mice (Bray et al., 1998). The MA-EBOV initially targets macrophages and other mononuclear phagocytes to invade the regional lymph nodes, similar to observations in humans, NHPs, and guinea pigs (Connolly et al., 1999; Davis et al., 1997; Gibb et al., 2001a, b; Zaki & Goldsmith, 1999). Thus,immunocompetent mouse models are considered ideal for studying fundamental aspects of viral replication, pathogenesis,and counter immune responses. They also provide an excellent platform for evaluating the therapeutic efficacy of candidate vaccines, antibodies, and small molecules such as anti-viral drugs (Kroeker et al., 2017a; Mendoza et al., 2016; Qiu et al.,2016; Zeitlin et al., 2016).
Marburg virus (MARV)
Marburg virus (MARV) and RAVV variants lethal to adult immunocompetent mice have been developed through sequential passage in the livers and spleens of severe,combined immunodeficient (SCID) mice (Warfield et al., 2007).Qiu et al. (2014) established a lethal mouse model against the Angolan strain MARV (MARV/Ang), the most virulent strain of MARV, with mice infected with 2 000 LD50of MARV/Ang-MA i.p.showing lymphopenia, thrombocytopenia, and uncontrolled viremia. Warfield et al. (2009) developed a lethal mouseadapted variant of RAVV (MA-RAVV), which caused death 5–10 days post infection (dpi), with disease manifestations including high viremia, fibrin degradation products, platelet loss,profound loss of tissue lymphocytes, and liver damage
lmmunocompromised Mouse Model
Ebola virus (EBOV)
Another method of studying WT filovirus infections in mice involves the use of mice with defects in the immune system.Bray et al. (2001) reported that i.p. inoculation of WT-EBOV was lethal to IFN-α/β receptor knockout (IFNAR–/–) mice, with immunohistochemical analysis demonstrating increased replicating virus and viral antigen in livers and spleens.Additionally, mice knocked out for transcription factor STAT1(which plays a key role in type I IFN signaling) were also highly susceptible to i.p. inoculation with 102pfu of WT-EBOV (Bray et al., 2001).
The type I IFN response plays a key role to resistance in mice. In addition, mouse models deficient in type I IFN signaling are more susceptible to WT filoviruses, serving an important model for studying pathogenesis. Immunodeficient mouse models of EBOV infection demonstrate high viremia and viral load in the spleen, liver, and multiple organ tissues.Lymphopenia, thrombocytopenia, kidney dysfunction, and liver damage have been observed in disease progression (Bradfute et al., 2007; Bradfute et al., 2010; Bray et al., 2001; Shurtleff &Bavari, 2015). As immunocompromised mice have a higher unit cost compared with immunocompetent mice, require handling in sterile conditions, and cannot mount a normal robust immune response (thus cannot determine immune correlates of protection or pathogenesis), they have not been used as extensively for research. However, the advantage to this animal model is that clinical WT filovirus strains can be used as a challenge virus to test the efficacy of candidate countermeasures rapidly without the need for host adaptation,which can be time consuming.
Marburg virus (MARV)
Warfield et al. (2007) examined SCID mice (which lack B and T cells) with intact IFN responses, who became sick when inoculated i.p. with ~103pfu of ‘SCID mouse-adapted’ MARVMusoke and died within 3–4 weeks after infection. However,after subsequent animal-to-animal passages, both RAVV and Musoke strains of MARV (MARV-Musoke and MARV-Ci67)caused rapidly lethal disease (Warfield et al., 2007). Initial signs of MARV disease in these animal models include fever,anorexia, rash, huddling, weight loss, dehydration, and diarrhea. Severe complications such as prostration, failure to respond to stimulation, hind limb paralysis, and bleeding from body orifices develop at 6–10 days. Early hematological and immunological changes include lymphopenia, neutrophilia, and profound thrombocytopenia, whereas alterations in serum chemistry levels, such as increases in liver enzymes, are also prominent after infection. At death, MARV was found at high titers in the blood, liver, spleen, kidneys, and other major organs, indicating systemic spread of the virus (Warfield et al.,2007).
Sudan virus (SUDV)
Mice deficient for type 1 IFN infections (α/β/γ IFN receptor knockout, IFN-α/βR?/?) have been found to be susceptible to wild-type SUDV (Brannan et al., 2015). Specifically, IFN-α/βR?/?mice challenged i.p. with a dose of 103pfu of SUDV Boneface variant (SUDV/Bon) subsequently exhibited ~25% weight loss,which peaked at 7–9 dpi. Maximum lethality was observed in the 103pfu challenge group, with a 63% survival, and mean time to death was approximately 11 days. Histological analyses of liver and spleen samples of the SUDV-infected mice on day 3 showed the development of hepatocellular degeneration and necrosis.
GUlNEA PlG MODEL
Ebola virus (EBOV)
Guinea pigs occupy a prominent role in filovirus research and are widely used for studying filoviral hemorrhagic fevers (Bowen et al., 1977; Cross et al., 2015a; Wong et al., 2015a, b, c).However, guinea pigs infected with WT filoviruses result in only transient illness (Ryabchikova et al., 1996; Simpson et al.,1968). Therefore, EBOV was serially passaged in the livers and spleens of guinea pigs until the adapted virus (GPA-EBOV)caused uniform lethality, with animals succumbing to death at 7–9 dpi (Simpson et al., 1968). At a dose of 103.8 pfu, GPAEBOV-infected animals showed fibrin deposition and thrombocytopenia during the late stages of infection (Connolly et al., 1999), as well as high viral titers in the spleen, liver,adrenal gland, and lungs. Viremia was observed in guinea pigs 2 days after inoculation, which peaked on day 7 (>104pfu/mL)(Connolly et al., 1999; Subbotina et al., 2010).
Guinea pigs infected with GPA-EBOV showed drastic histopathological changes in Kupffer cells (especially virus replication), death of hepatocytes, and destruction of the sinusoid wall (Connolly et al., 1999), with the spleen and lymph nodes demonstrating lymphoid necrosis along with neutrophilia and lymphopenia (Connolly et al., 1999; Subbotina et al., 2010).Remarkably, lymphocyte bystander apoptosis was not prominent in these animals (Bradfute et al., 2007; Bray et al.,1998; Connolly et al., 1999). While the guinea pig model recapitulates some aspects of human filoviral disease, its higher unit cost, need for more specialized housing/husbandry requirements, and the lack of reagents to characterize aspects of innate/adaptive immune responses (such as T-cell immunity or cytokine levels) mean that it is more suited as a secondary animal model for confirming experimental results and trends from mice studies.
Adaptation of MARV (isolates Ci67 and Musoke) in guinea pigs from previous research resulted in their deaths on days 7–9, with body temperatures recorded at 41.1 °C (Simpson et al.,1968) and clinical symptoms such as bloated face and loss of appetite and weight evident. Cross et al. (2015b) evaluated and compared pathogenicity in outbred guinea pigs with GPARAVVUTMBand GPA-MARV infections to better understand the disease pathogenesis of MARV infection in the guinea pig model. Results demonstrated prolonged clotting times (PT and APTT) associated with an increase in plasminogen activator inhibitor 1 and von Willebrand factor activity in GPA-RAVV and GPA-MARV-Angola-infected animals. In addition, circulating protein C activity and tissue factor concentrations also decreased during the time of death.
Marburg virus (MARV)
Guinea pigs infected with approximately 5×103pfu of GPAMARV-Angola showed remarkable differences in tissue factor kinetics (Cross et al., 2015b). In these animals, prolonged clotting times (PT and APTT) were associated with an increase in plasminogen activator inhibitor 1 and von Willebrand factor activity. Circulating protein C activity and tissue factor concentrations were also decreased at death. Thus, these proteins likely played a significant role in coagulopathy.Furthermore, disturbance of the fibrinolysis pathway was implicated by the depression in the metabolism of bradykinin,prekallikrein, and thrombin-activated fibrinolysis inhibitor (TAFI).Interestingly, GPA-RAVV-infected animals displayed increased TAFI concentrations at 3 dpi, whereas GPA-MARV-Angolainfected animals showed a decrease in TAFI concentrations at late stage infection. Inflammatory mediators such as TNF-a, IL-6, nitric oxide, and HMGB-1 were also elevated during the late stages of infection.
Sudan virus (SUDV)
Wong et al. (2015a) developed and characterized a guinea pig-adapted SUDV variant based on the Boneface variant through serial passaging of WT-SUDV in the livers and spleens, and showed that the adapted virus caused uniform lethality in guinea pigs. The guinea pig-adapted SUDV variant (SUDV-GA) caused LD50to animals at a dose of 5.3×10-250% tissue culture infective doses (TCID50).Animals infected with SUDV-GA showed high viremia and died between 9–14 dpi. Several hallmarks of SUDV infection were observed, such as lymphadenopathy, elevated liver enzyme activities, and coagulation abnormalities.Histopathology and immunohistochemistry findings indicated that the SUDV-GA antigen was present in the livers and spleens of infected animals.
SYRlAN HAMSTER MODEL
Ebola virus (EBOV)
A Syrian golden hamster model for EBOV was developed by inoculating 6-week-old hamsters i.p. with 103focus forming units (ffu) of MA-EBOV (Ebihara et al., 2013; Zivcec et al.,2011). Hamsters i.p. inoculated with MA-EBOV (Bray et al.,1998) showed signs of disease, such as ruffled fur and decreased activity beginning at 3 dpi, and all animals died by days 4–5, with correspondingly high virus titers in the spleen,liver, kidneys, heart, lungs, and brain, indicating systemic spread of the virus (Ebihara et al., 2013).
Viremia was detected beginning on day 2 and reached ~108pfu/mL by day 4, with infected hamsters showing rapid and increased distribution of the viral antigen in the spleen and liver(Ebihara et al., 2013). In the spleen, macrophages and marginal reticular-like cells in the red pulp and marginal zone showed higher virus replication. In the liver, Kupffer cells were the first target cells for MA-EBOV, which then spread to hepatocytes starting at 2 dpi. Importantly, coagulopathy was observed in the MA-EBOV-infected Syrian hamsters, but was absent in other rodent models. In addition, thrombocytopenia and decreased protein C concentrations were also noted in the infected hamsters. Using in-house primer-probe sets, Ebihara et al. (2013) also showed that MA-EBOV infection induced the expression of cytokine/chemokine genes, including IL-1b, IL-6,TNF-a, IL-12p35, IP-10, and IL-10, in the spleen and liver,indicating an uncontrolled immune response. Due to the presence of rash and induction of cytokines/chemokines, the Syrian hamster more closely recapitulates EBOV disease compared with guinea pigs; however, the higher unit costs,need for specialized housing/husbandry equipment, and lack of commercial reagents for studying innate and cell-mediated immune responses means that this model is still not widely used in filovirus research.
Marburg virus (MARV)
Marzi et al. (2016) recently developed a novel Syrian golden hamster model for MARV infection using a hamster-adapted MARV variant Angola (HA-MARV). This model provides new insight on virus pathogenesis in hamsters, and moreover closely resembles MARV infection in humans and NHPs,including hemorrhagic manifestations and coagulation abnormalities. In this study, hamsters were infected i.p. with 10–1–103pfu of HA-MARV, leading to significant weight loss and disease surrender between 7–11 dpi. The HA-MARV infected hamsters displayed an increase in body temperature at 6 dpi and drop in temperature during the end stage of the disease. In contrast, the mock-infected animals (WT-MARV) did not show any changes in body temperature or weight, suggesting that infection with WT-MARV failed to induce the disease in hamsters. Maculopapular rashes along with visible petechial hemorrhages on the face and abdomen were noted in the HAMARV-infected hamsters on 7 dpi. Hemorrhage was also identified in the footpads and joints, as well as in the kidneys.Coagulation parameters in HA-MARV-infected hamsters such as PT, APTT, and thrombin increased with delay in blood clot formation during the infection. However, the animals infected with WT-MARV did not exhibit these coagulation abnormalities.The HA-MARV replication was found to be robust, with viremia peaking at 108TCID50/mL in the blood, 107TCID50/mL in the liver, 106TCID50/mL in the spleen, and 105TCID50/mL in the mesenteric lymph nodes by 7–8 dpi. Dysregulated immune responses were also observed in hamsters infected with HAMARV; for example, pro-inflammatory chemokines MIP-1α and IP-10, as well as type I interferon responses, were elevated when compared to WT-MARV-infected animals.
FERRET MODEL
Ebola virus (EBOV)
The susceptibility of domestic ferrets (Mustela putorius furo) to EBOV has been studied recently. Cross et al. (2016)demonstrated that ferrets infected intranasally (i.n.) with the EBOV Kikwit variant at 103pfu, showed uncontrolled viral replication, abnormalities in white blood cell counts, and multiple-organ failure. Initial signs of the disease in the EBOV-infected ferrets were first observed at 3 dpi, followed by rapidonset hypothermia and weight loss on day 4. Remarkably,ferrets died between 6–9 dpi. Necropsy inspection revealed pathological features of hemorrhagic fever, including petechial rashes on the skin surface, reticulated pallor of the liver, and mottled splenomegaly. All ferrets showed progressive neutrophilia and lymphocytopenia beginning on 4 dpi. Vascular leakage was also noted with hypoalbuminemia and hypoproteinemia, and increasing levels of circulating proinflammatory markers TNF-α and nitric oxide were also recorded on day 4.
Kozak et al. (2016) also reported on a ferret model for EBOV.Animals were infected with 200 TCID50via the i.m or i.n route,and showed peak viremia of 107GEQ/mL or 109TCID50/mL by 5 or 6 dpi, with evidence of sporadic viral shedding from the oral, nasal, and rectal cavities. Notably, weight loss was less pronounced in animals infected with EBOV compared with BDBV-infected animals from the same study. Biochemical analyses further demonstrated increases in concentrations/activities of ALT, ALP, and BIL, suggesting liver damage, and increases in BUN and CRE, indicating elevated renal problems.Prolonged APTT and thrombin time (TT), along with an increase in fibrinogen levels, were also noted, suggesting that ferrets recapitulated disseminated intravascular coagulation.Interestingly, EBOV-infected ferrets displayed decreased levels of serum albumin after EBOV challenge, which might be the cause of disease-induced edema.
The results from the above studies demonstrate that ferrets are highly susceptible to EBOV infection and the pathological parameters observed during disease pathogenesis closely resemble those found in humans.
Sudan virus (SUDV)
Kroeker et al. (2017b) demonstrated that ferrets are susceptible to wild-type SUDV infections. Ferrets inoculated with WT-SUDV via i.m and i.n experienced viremia, organ dysfunction, viral shedding, and eventual death. A decrease in white blood cell counts, increase in fibrinogen, and decrease in platelets and PT%, which are indicative symptoms of disseminated intravascular coagulopathy, were observed further during disease progression. Cross et al. (2016) also described a lethal model of SUDV with the domestic ferret. Ferrets were challenged i.n. with 103pfu of SUDV, with initial signs of the disease observed at 3 dpi and rapid-onset hypothermia and weight loss detected by day 4 for SUDV-infected ferrets. Clinical signs included diarrhea, dehydration, nasal and ocular discharge,labored breathing, hunched posture, and altered gait, with death occurring at 6–9 dpi. Hematoxylin-eosin staining revealed that the most significant lesions in the infected ferrets were lymphohistiocytic and neutrophilic necrotizing hepatitis and necrotizing splenitis.
Bundibugyo virus (BDBV)
Kozak et al. (2016) challenged ferrets with 159 TCID50of BDBV via the i.m. route, and observed viremia at 4 dpi and uniform lethality at 8 dpi. In all BDBV-infected animals, significant increases in coagulation parameters were observed, including in APTT, TT, and PT. Hematological analysis revealed a decrease in white blood cell counts, lymphocytes, and platelets in ferrets after infection. Petechial rashes were also observed during the disease.
NON-HUMAN PRlMATE (NHP) MODELS
Ebola virus (EBOV)
Many reports have demonstrated African green monkeys(Chlorocebus aethiops), cynomolgus macaques (Macaca fascicularis), and rhesus macaques (Macaca mulatta) as excellent models for EBOV infection (Bowen et al., 1978; Davis et al., 1997; Ellis et al., 1978; Fisher-Hoch et al., 1992; Geisbert et al., 2003a, b, c, d; Ignatiev et al., 2000; Jaax et al., 1996;Jahrling et al., 1996; Johnson et al., 1995; Ryabchikova et al.,1999; Wong et al., 2016). Following EBOV infection, NHPs became febrile (above 40 °C) at 3 dpi, with pyrexia and temperature decrease exhibited throughout the course of the disease, followed by death within 5–8 dpi (Baskerville et al.,1978; Bowen et al., 1978; Ellis et al., 1978; Fisher-Hoch et al.,1985; Luchko et al., 1995). Viremia has been observed at 3 dpi with titers reaching 106.5?107pfu/mL on days 4–5 (Bowen et al.,1978; Fisher-Hoch et al., 1992; Jahrling et al., 1996). Compared with other NHP species, cynomolgus and rhesus macaques are the best available animal models due to their susceptibility to EBOV infections (Bente et al., 2009), thus providing a stringent test for any candidate medical countermeasure. In addition,African green monkeys and baboons infected with EBOV show impairment of the coagulation system. For example, fibrin thrombosis was localized to all visceral organs in African green monkeys, whereas hemorrhage was most prominent in the visceral organs such as the liver and spleen in baboons(Ignatiev et al., 2000; Ryabchikova et al., 1999).
Currently, macaques well recapitulate pathological features of fatal filovirus disease observed in humans, including high viremia, coagulation abnormalities such as decreased platelet levels and increased blood clotting times, and aberrant proinflammatory cytokine responses, including the release of IL-6 and MCP-1 (Geisbert et al., 2003a, 2015). Studies in macaques have also demonstrated that doses of EBOV as low as 2?15 pfu administered by different challenge routes can produce lethal filovirus infection (Sullivan et al., 2000, 2003).Ebola virus disease (EVD) signs in macaques initially occur at 3?5 days after exposure, with symptoms including fever and malaise, followed by anorexia, depression, lethargy, diarrhea,vomiting, and maculopapular rash. Hemorrhagic manifestations have also been identified, and include petechiae, ecchymosis,and bruising, hemorrhage at venipuncture sites, epistaxis,hematochezia, and hematuria. In addition, neutrophilia,lymphopenia, thrombocytopenia, and early monocytosis indicate abnormalities in complete blood count parameters.
Although airborne transmission is not considered a significant route of human infection (Osterholm et al., 2015), aerosolized viruses have caused lethal disease in experimentally-infected NHPs. Previous study showed that control rhesus macaques,located 3 m from experimental rhesus macaques inoculated i.m. with EBOV, became infected (Jaax et al., 1995), with antigen staining patterns of pulmonary specimens implicating aerosol infection. Conversely, other research has shown no transmission between rhesus macaques infected i.m. with EBOV and uninfected rhesus macaques when housed adjacent to each other (Alimonti et al., 2014). However, virus transmission from pigs infected oro-nasally with EBOV to na?ve NHPs without direct contact was shown to be possible, with the infected NHPs exhibiting extensive lung damage (Weingartl et al., 2012). Mucosal exposure as a source of infection in rhesus macaques (through conjunctival and oral routes), required higher doses (5.2 log10of EBOV Mayinga isolate) compared to parenteral routes; however, doses of 10 pfu by oral or conjunctival routes did not result in any clinical disease (Mire et al., 2013). Thus, despite the above studies, an aerosol model of EBOV infection in NHPs consistently causing 100% lethality has not yet been established.
Marburg virus (MARV)
NHPs are susceptible to MARV infections and have been extensively used to study its pathogenesis. Fernando et al.(2015) reported that cynomolgus macaques showed severe disease against Marburg Angola compared with other MARV strains, with high fever, anorexia, and lymphopenia, followed by death (mean time after infection of 6.7 days). Peak viremia reached 104–107TCID50/mL by 4 dpi. Cynomolgus macaques showed febrile illness, anorexia, diarrhea, skin rash, and hemorrhagic manifestations at 2–6 dpi following high dose (103pfu) infection by MARV Angola (Alves et al., 2010; Geisbert et al., 2007; Hensley et al., 2011; Simpson et al., 1968; Simpson,1969; Murphy et al., 1971), with a sudden decrease in body temperature followed by death at 6–13 dpi. Lymphocytosis was observed at the beginning of the illness (Geisbert et al., 2007;Gonchar et al., 1991; Johnson et al., 1996; Simpson et al., 1968;Simpson, 1969; Spiridonov et al., 1992), whereas thrombocytopenia and leukocytosis with increased neutrophilia were observed 5–6 dpi (Hensley et al., 2011). The infection was also observed to spread to the liver, adrenal glands, and finally to endothelial cells in a variety of organ tissues (Hensley et al., 2011). Viremia occurred on day 3 in cynomolgus macaques and African green monkeys, reaching peak titers of 107?108pfu/mL at 8 dpi(Hensley et al., 2011).
Sudan virus (SUDV)
SUDV infection has been shown to cause death in the NHP model (rhesus macaques) 7?10 dpi (Thi et al., 2016).Specifically, adult rhesus macaques inoculated i.m. with 103pfu of SUDV Gulu succumbed to infection after 7–10 days, with viremia peaking at 108pfu/mL at 7 dpi, accompanied by liver and renal dysfunction as evidenced by increased ALT, GGT,BUN, and CRE levels. Control and treated animals that succumbed to SUDV infection showed lesions (observed by hematoxylin and eosin staining) in tissues. Significant lesions included splenic lymphoid depletion with tangible body macrophages and fragmented nuclear debris and enlargement of splenic red pulp with fibrin, multifocal necrotizing hepatitis with sinusoidal leukocytosis, and mild interstitial pneumonia.Furthermore, depression, anorexia, petechial rash, and hemorrhage were observed in the animals infected with SUDV.
Bundibugyo virus (BDBV)
Cynomolgus macaques have been used for studying the pathogenesis of BDBV infection (Mire et al., 2013), in which macaques were challenged i.m. with 103pfu of BDBV. Animals were monitored for clinical signs of illness over the course of 28 days. Clinical scores were recorded each day post-challenge for each animal using a scoring system based on dyspnea,depression, recumbency, and rash. No antibody titers (IgG) for the control group were observed against BDBV. The signs of disease in response to BDBV infection were more dramatic for the control animals. Signs observed between day 0?28 after BDBV challenge included fever, anorexia, depression,rectorrhagia, lymphopenia, and thrombocytopenia. Serum concentrations of alkaline phosphatase (ALP), aspartate aminotransferase (AST), blood urea nitrogen (BUN), and gamma glutamyltransferase (GGT) were elevated, and by days 10?11 all animals had died.
Tai Forest virus (TAFV)
TAFV may cause severe disease and death in NHPs, as evidenced by the severe infection of one person after coming into contact with a dead chimpanzee, although the human patient ultimately survived (Guenno et al., 1995). To date, there have been no other reported infections, and thus this virus has not been the subject of extensive studies and there is a lack of animal models.
SUMMARY
Animal models are important for the study of filovirus pathogenesis, with the goal of testing the effectiveness of specific antivirals and vaccines. A variety of animal species are susceptible to WT and host-adapted filoviruses, and are thus used to test the efficacy of anti-filovirus products during nonoutbreak situations. The Food and Drug Administration’s “twoanimal rule” states that the efficacy of a candidate product should be tested in “more than one animal species expected to react with a response predictive for humans” (FDA, 2017).Therefore, any vaccine or antiviral must demonstrate safety and efficacy in at least two animal models (a smaller animal model in addition to a NHP model) to have any hope of being licensed.
The most important aspects of filovirus infection are summarized in Table 3. Parameters such as lymphopenia, liver damage, thrombocytopenia, coagulopathy, cytokine storm,rash, and hemorrhage are the chief hallmarks of filoviral diseases. Due to their advantages in terms of handling, cost,and access, mice and guinea pigs often serve as primary and secondary animal models for preliminary experiments and diverse genetic studies (such as knockout mice); however, as viruses can adapt to hosts and each host cannot recapitulate all hallmarks of filovirus diseases, mice and guinea pigs have limited translatability and predictability to human filoviral disease and thus are mostly used to screen potentially efficacious compounds. Currently, NHPs are the only animal species available that can recapitulate all stated aspects and thus are considered as the gold standard. Ferrets are the newest animal model to gain attention in filovirus research (Cross et al., 2016;Kozak et al., 2016; Kroeker et al., 2017b). In addition to being small, ferrets can accurately recapitulate human filoviral diseases, including coagulopathy. Syrian golden hamsters can also recapitulate disease that closely mimics that in humans and NHPs, including coagulopathy. However, this model suffers from the need for host-adapted viruses and the lack of widespread reagents for studying immune responses.Therefore, ferrets and NHPs are likely the two best animal models at present for the testing of anti-filovirus compounds.
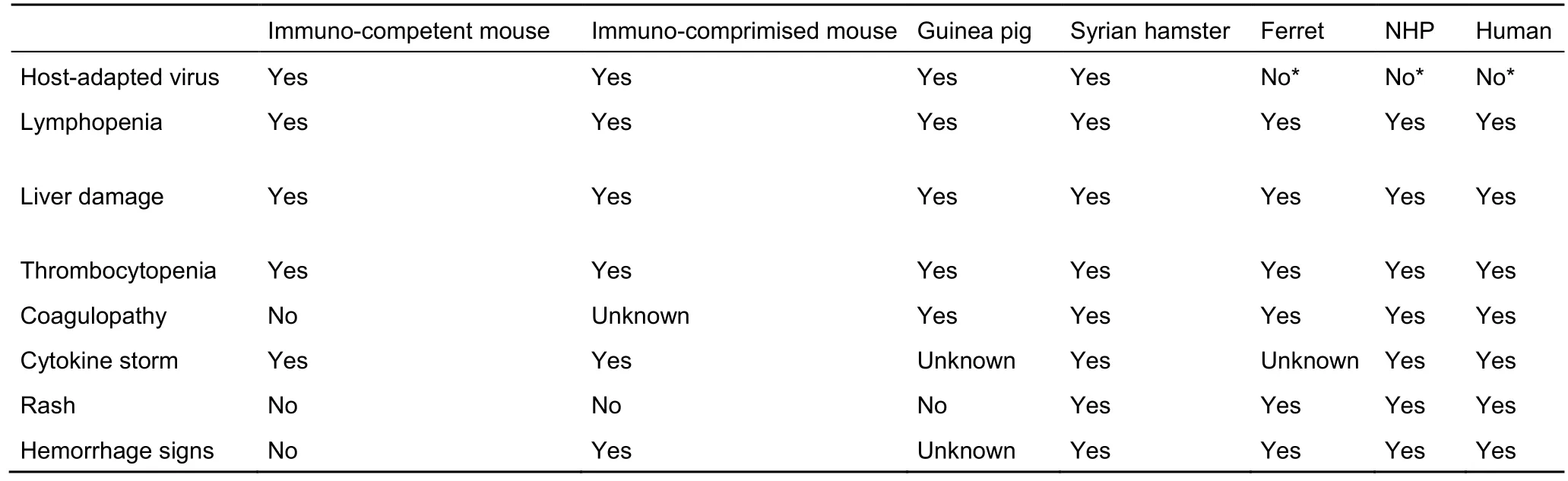
Table 3 Clinical manifestations in different animal models of filovirus infections
While this review has covered the different animal models available for each filovirus (Table 1), it is important to note that models against some viruses are more developed than others.There is an abundance of EBOV animal models, but very limited models available against TAFV. In addition, only ferrets have been used as a small animal model for BDBV. Immediate future goals should be the development and characterization of rodent animal models for the lesser known filoviruses. With experience gained from previous studies and well-established methods, the goal of establishing the necessary animal models for each known filovirus is attainable.
COMPETlNG lNTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRlBUTlONS
V.S. wrote the manuscript. G.W. and X.Q. revised the manuscript. All authors read and approved the final manuscript.
Alimonti J, Leung A, Jones S, Gren J, Qiu XG, Fernando L, Balcewich B,Wong G, Str?her U, Grolla A, Strong J, Kobinger G. 2014. Evaluation of transmission risks associated with in vivo replication of several high containment pathogens in a biosafety level 4 laboratory. Scientific Reports,4: 5824.
Alves DA, Glynn AR, Steele KE, Lackemeyer MG, Garza NL, Buck JG,Mech C, Reed DS. 2010. Aerosol exposure to the angola strain of marburg virus causes lethal viral hemorrhagic fever in cynomolgus macaques.Veterinary Pathology, 47(5): 831–851.
Banadyga L, Dolan MA, Ebihara H. 2016. Rodent-adapted filoviruses and the molecular basis of pathogenesis. Journal of Molecular Biology, 428(17):3449–3466.
Baskerville A, Bowen ETW, Platt GS, McArdell LB, Simpson DIH. 1978.The pathology of experimental ebola virus infection in monkeys. Journal of Pathology, 125(3): 131–138.
Bente D, Gren J, Strong JE, Feldmann H. 2009. Disease modeling for ebola and marburg viruses. Disease Models & Mechanisms, 2(1–2): 12–17.Bowen ET, Lloyd G, Harris WJ, Platt GS, Baskerville A, Vella E. 1977. Viral haemorrhagic fever in southern sudan and northern zaire. Preliminary studies on the aetiological agent. Lancet, 1(8011): 571–573.
Bowen ETW, Platt GS, Simpson DIH, McArdell LB, Raymond RT. 1978.Ebola haemorrhagic fever: experimental infection of monkeys. Transactions of the Royal Society in Tropical Medicine and Hygiene, 72(2): 188–191.
Bradfute SB, Braun DR, Shamblin JD, Geisbert JB, Paragas J, Garrison A,Hensley LE, Geisbert TW. 2007. Lymphocyte death in a mouse model of ebola virus infection. Journal of Infectious Disease, 196(S2): S296-S304.
Bradfute SB, Swanson PE, Smith MA, Watanabe E, McDunn JE, Hotchkiss RS, Bavari S. 2010. Mechanisms and consequences of ebolavirus-induced lymphocyte apoptosis. Journal of Immunology, 184(1): 327–335.
Bradfute SB, Warfield KL, Bray M. 2012. Mouse models for filovirus infections. Viruses, 4(9): 1477–1508.
Brannan JM, Froude JW, Prugar L, Bakken RR, Zak SE, Daye SP,Wilhelmsen CE, Dye JM. 2015. Interferon α/β receptor-deficient mice as a model for ebola virus disease. Journal of Infectious Diseases, 212(S2):S282–S294.
Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. 1998. A mouse model for evaluation of prophylaxis and therapy of ebola hemorrhagic fever.Journal of Infectious Diseases, 178(3): 651–661.
Bray M. 2001. The role of the type I interferon response in the resistance of mice to filovirus infection. Journal of General Virology, 82(Pt 6): 1365–1373.Bray M, Hatfill S, Hensley L, Huggins JW. 2001. Haematological,biochemical and coagulation changes in mice, guinea-pigs and monkeys infected with a mouse-adapted variant of ebola zaire virus. Journal of Comparative Pathology, 125(4): 243–253.
Connolly BM, Steele KE, Davis KJ, Geisbert TW, Kell WM, Jaax NX,Jahrling PB. 1999. Pathogenesis of experimental ebola virus infection in guinea pigs. Journal of Infectious Diseases, 179(S1): S203-S217.
Cross RW, Fenton KA, Geisbert JB, Mire, ME, Geisbert TW. 2015a.Modeling the disease course of zaire ebolavirus infection in the outbred guinea pig. Journal of Infectious Diseases, 212(S2): S305–S315.
Cross RW, Fenton KA, Geisbert JB, Ebihara H, Mire CE, Geisbert TW.2015b. Comparison of the pathogenesis of the Angola and Ravn strains of Marburg virus in the outbred guinea pig model. The Journal of Infectious Diseases, 212(S2): S258–S270.
Cross RW, Mire CE, Borisevich V, Geisbert JB, Fenton KA, Geisbert TW.2016. The domestic ferret (mustela putorius furo) as a lethal infection model for 3 species of ebolavirus. Journal of Infectious Diseases, 212(S4):565–569.
Davis KJ, Anderson AO, Geisbert TW, Steele KE, Geisbert JB, Vogel P,Connolly BM, Huggins JW, Jahrling PB, Jaax NK. 1997. Pathology of experimental ebola virus infection in african green monkeys. Involvement of fibroblastic reticular cells. Archives of Pathology & Laboratory Medicine,121(8): 805–819.
Ebihara H, Takada A, Kobasa D Jones S, Neumann G, Theriault S, Bray M,Feldmann H Kawaoka Y. 2006. Molecular determinants of ebola virus virulence in mice. PLoS Pathogens, 2(7): e73.
Ebihara H, Zivcec M, Gardner D, Falzarano D, LaCasse R, Rosenke R,Long D, Haddock E, Fischer E, Kawaoka Y, Feldmann H. 2013. A Syrian golden hamster model recapitulating Ebola hemorrhagic fever. The Journal of Infectious Diseases, 207(2): 306–318.
Ellis DS, Bowen ET, Simpson DI, Stamford S. 1978. Ebola virus: a comparison, at ultrastructural level, of the behaviour of the sudan and zaire strains in monkeys. British Journal of Experimental Pathology, 59(6): 584–593.
Fernando L, Qiu X, Melito PL, Williams KJ, Feldmann F, Feldman H, Jones SM, Alimonti JB. 2015. Immune response to marburg virus angola infection in nonhuman primates. Journal of Infectious Disease, 212(S2): S234–S241.Faye O, Faye O, Soropogui B, Patel P, El Wahed AA, Loucoubar C, Fall G,Kiory D, Magassouba N, Keita S, Kondé MK, Diallo AA, Koivogui L,Karlberg H, Mirazimi A, Nentwich O, Piepenburg O, Niedrig M, Weidmann M, Sall AA. 2015. Development and deployment of a rapid recombinase polymerase amplification Ebola virus detection assay in Guinea in 2015.Euro Surveillance, 20(44), doi: 10.2807/1560–7917.ES.2015.20.44.30053.FDA. gov. 2017. CFR - Code of Federal Regulations Title 21.https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=601.91
Fisher-Hoch SP, Platt GS, Neild GH, Southee T, Baskerville A, Raymond RT, Lloyd G, Simpson DI. 1985. Pathophysiology of shock and hemorrhage in a fulminating viral infection (ebola). Journal of Infectious Diseases,152(5): 887–894.
Fisher-Hoch SP, Brammer TL, Trappier SG, Hutwagner LC, Farrar BB, Ruo SL, Brown BG, Hermann LM, Perez-Oronoz GI, Goldsmith CS, Hanes MA,McCormick JB. 1992. Pathogenic potential of filoviruses: role of geographic origin of primate host and virus strain. Journal of Infectious Diseases,166(4): 753–763.
Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J,Young HA, Fredeking WE, Rote WE, Vlasuk GP. 2003a. Treatment of ebola virus infection with a recombinant inhibitor of factor viia/tissue factor: a study in rhesus monkeys. The Lancet, 362(9400): 1953–1958.
Geisbert TW, Hensley LE, Larsen T, Young HA, Reed DS, Geisbert, JB,Scott DP, Kagan E, Jahrling PB, Davis KJ. 2003b. Pathogenesis of ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. American Journal of Pathology,163(6): 2347–2370.
Geisbert TW, Young HA, Jahrling PB, Davis KJ, Kagan E, Hensley LE.2003c. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. Journal of Infectious Diseases, 188(11):1618–1629.
Geisbert TW, Young HA, Jahrling PB, Davis KJ, Larsen T, Kagan E,
Hensley LE. 2003d. Pathogenesis of ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. American Journal of Pathology, 163(6): 2371–2382.
Geisbert TW, Daddario-DiCaprio KM, Geisbert JB, Young HA, Formenty P,Fritz EA, Larsen T, Hensley LE. 2007. Marburg virus angola infection of rhesus macaques: pathogenesis and treatment with recombinant nematode anticoagulant protein c2. Journal of Infectious Diseases, 196(S2): S372–S381.
Geisbert TW, Strong JE, Feldmann H. 2015. Considerations in the use of nonhuman primate models of Ebola virus and Marburg virus infection.Journal of Infectious Diseases, 212(S2): S91–S97.
Gibb TR, Norwood Jr DA, Woollen N, Henchal EA. 2001a. Development and evaluation of a fluorogenic 5' nuclease assay to detect and differentiate between ebola virus subtypes zaire and sudan. Journal of Clinical Microbiology, 39(11): 4125–4130.
Gibb TR, Bray M, Geisbert TW, Steele KE, Kell WM, Davis KJ, Jaax NX.2001b. Pathogenesis of experimental ebola zaire virus infection in BALB/c mice. Journal of Comparative Pathology, 125(4): 233–242.
Gonchar NI, Pshenichnov VA, Pokhodiaev VA, Lopatov KL, Firsova IV.1991. The sensitivity of different experimental animals to the marburg virus.Voprosy Virusologii, 36(5): 435–437.
Hensley LE, Alves DA, Geisbert JB, Fritz EA, Reed C, Larsen T, Geisbert TW. 2011. Pathogenesis of marburg hemorrhagic fever in cynomolgus macaques. Journal of Infectious Diseases, 204(S3): S1021-S1031.
Ignatiev GM, Dadaeva AA, Luchko SV, Chepurnov AA. 2000. Immune and pathophysiological processes in baboons experimentally infected with ebola virus adapted to guinea pigs. Immunology Letters, 71(2): 131–140.
Jaax N, Jahrling P, Geisbert T, Geisbert J, Steele K, McKee K, Nagley D,Johnson E, Jaax G, Peters C. 1995. Transmission of Ebola virus (Zaire strain) to uninfected control monkeys in a biocontainment laboratory. The Lancet, 346(8991–8992): 1669–1671.
Jaax NK, Davis KJ, Geisbert TJ, Vogel P, Jaax GP, Topper M Jahrling PB.1996. Lethal experimental infection of rhesus monkeys with ebola-zaire(mayinga) virus by the oral and conjunctival route of exposure. Archives of Pathology & Laboratory Medicine, 120(2): 140–155.
Jahrling PB, Geisbert TW, Jaax NK, Hanes MA, Ksiazek TG, Peters CJ.1996. Experimental infection of cynomolgus macaques with ebola-reston filoviruses from the 1989–1990 U.S. Epizootic. Archives of Virology.Supplementum, 11: 115–134.
Johnson E, Jaax N, White J, Jahrling P. 1995. Lethal experimental infections of rhesus monkeys by aerosolized ebola virus. International Journal of Experimental Pathology, 76(4): 227–236.
Johnson ED, Johnson BK, Silverstein D, Tukei P, Geisbert TW, Sanchez AN, Jahrling PB. 1996. Characterization of a new marburg virus isolated from a 1987 fatal case in kenya. Archives of Virology. Supplementum, 11:101–114.
Kozak R, He SH, Kroeker A, de La Vega MA, Audet J, Wong G, Urfano C,Antonation K, Embury-Hyatt C, Kobinger GP, Qiu XG. 2016. Ferrets infected with bundibugyo virus or ebola virus recapitulate important aspects of human filovirus disease. Journal of Virology, 90(20): 9209–9223.
Kroeker A, Griffin BD, Qiu XG, Kobinger G. 2017a. Assessing antiviral countermeasures using mouse models of ebolavirus infection. In: Hoenen T,Groseth A. Ebolaviruses. New York, NY: Humana Press, 1628: 273–282.
Kroeker A, He SH, de La Vega MA, Wong G, Embury-Hyatt C, Qiu XG.2017b. Characterization of Sudan ebolavirus infection in ferrets. Oncotarget,8(28): 46262–46272.
Kuhn JH, Dodd LE, Wahl-Jensen V, Radoshitzky SR, Bavari S, Jahrling PB.2011. Evaluation of perceived threat differences posed by filovirus variants.Biosecurity and Bioterrorism, 9(4): 361–371.
Kuhn JH, Bao YM, Bavari S, Becker S, Bradfute S, Brister JR, Bukreyev AA,Chandran K, Davey RA, Dolnik O, Dye JM, Enterlein S, Hensley LE, Honko AN, Jahrling PB, Johnson KM, Kobinger G, Leroy EM, Lever MS,Mühlberger E, Netesov SV, Olinger GG, Palacios G, Patterson JL, Paweska JT, Pitt L, Radoshitzky SR, Saphire EO, Smither SJ, Swanepoel R, Towner JS, van der Groen G, Volchkov VE, Wahl-Jensen V, Warren TK, Weidmann M, Nichol ST. 2013. Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family filoviridae. Archives of Virology, 158(1): 301–311.
Le Guenno B, Formenty P, Wyers M, Gounon P, Walker F, Boesch C. 1995.Isolation and partial characterisation of a new strain of Ebola virus. The Lancet, 345(8960): 1271–1274.
Leroy EM, Epelboin A, Mondonge V, Pourrut X, Gonzalez JP, Muyembe-Tamfum JJ, Formenty P. 2009. Human ebola outbreak resulting from direct exposure to fruit bats in luebo, democratic republic of congo. 2007. Vector-Borne and Zoonotic Diseases, 9(6): 723–728.
Luchko SV, Dadaeva AA, Ustinova EN, Sizikova LP, Riabchikova EI,Sandakhchiev LS. 1995. Experimental study of ebola hemorrhagic fever in baboon models. Biulleten' Eksperimental'noi Biologii i Meditsiny, 120(9):302–304.
Martin B, Hoenen T, Canard B, Decroly E. 2016. Filovirus proteins for antiviral drug discovery: a structure/function analysis of surface glycoproteins and virus entry. Antiviral Research, 135: 1–14.
Marzi A, Banadyga L, Haddock E, Thomas T, Shen K, Horne EJ, Scott DP,Feldmann H, Ebihara H. 2016. A hamster model for Marburg virus infection accurately recapitulates Marburg hemorrhagic fever. Scientific Reports, 6:39214.
Mendoza EJ, Qiu XG, Kobinger GP. 2016. Progression of ebola therapeutics during the 2014–2015 outbreak. Trends in Molecular Medicine,22(2): 164–173.
Mire CE, Geisbert JB, Marzi A, Krystle N, Agans KN, Feldmann H, Geisbert TW. 2013. Vesicular stomatitis virus-based vaccines protect nonhuman primates against Bundibugyo ebolavirus. PLoS Neglected Tropical Diseases, 7(12): e2600.
Murphy FA, Simpson DI, Whitfield SG, Zlotnik I, Carter GB. 1971. Marburg virus infection in monkeys. Ultrastructural studies. Laboratory Investigation,24(4): 279–291.
Nakayama E, Saijo M. 2013. Animal models for Ebola and Marburg virus infections. Frontiers in Microbiology, 4: 267.
Osterholm MT, Moore KA, Kelley NS, Brosseau LM, Wong G, Murphy FA,Peters CJ, LeDuc JW, Russell PK, Van Herp M, Kapetshi J, Muyembe JJT,Ilunga BK, Strong J, Grolla A, Wolz A, Kargbo B, Kargbo DK, Formenty P,Sanders DA, Kobinger GP. 2015. Transmission of Ebola viruses: what we know and what we do not know. mBio, 6(2): e00137–15.
Qiu XG, Wong G, Audet J, Cutts T, Niu YL, Booth S, Kobinger GP. 2014.Establishment and characterization of a lethal mouse model for the angola strain of marburg virus. Journal of Virology, 88(21): 12703–12714.
Qiu XG, Audet J, Lv M, He SH, Wong G, Wei HY, Luo LL, Fernando L,Kroeker A, Bovendo HF, Bello A, Li F, Ye P, Jacobs M, Ippolito G, Saphire EO, Bi S, Shen B, Gao GF, Zeitlin L, Feng J, Zhang B, Kobinger GP. 2016.Two-mab cocktail protects macaques against the makona variant of ebola virus. Science Translational Medicine, 8(329): 329ra333.
Ryabchikova E, Kolesnikova L, Smolina M, Tkachev V, Pereboeva L,Baranova S, Grazhdantseva A, Rassadkin Y. 1996. Ebola virus infection in guinea pigs: presumable role of granulomatous inflammation in pathogenesis. Archives of Virology, 141(5): 909–921.
Ryabchikova EI, Kolesnikova LV, Luchko SV. 1999. An analysis of features of pathogenesis in two animal models of ebola virus infection. Journal of Infectious Diseases, 179(S1): S199–S202.
Sanchez A, Geisbert TW, Felsmann H. 2007. Filoviridae: Marburg and Ebola viruses. In: Knipe DM, Howley PM. Fields Virology. Philadelphia, PA:Lippincott Williams and Wilkins.
Shurtleff AC, Bavari S. 2015. Animal models for ebolavirus countermeasures discovery: What defines a useful model? Expert Opinion on Drug Discovery, 10(7): 685–702.
Simpson DI, Zlotnik I, Rutter DA. 1968. Vervet monkey disease. Experiment infection of guinea pigs and monkeys with the causative agent. British Journal of Experimental Pathology, 49(5): 458–464.
Simpson DIH. 1969. Marburg agent disease: in monkeys. Transactions of the Royal Society of Tropical Medicine and Hygiene, 63(3): 303–309.
Spiridonov VA, Bazhutin NB, Belanov EF, Vo?tenko AV, Zolin VV,Krivenchuk NA, Omel'chenko NI, Polikanov VP, Tereshchenko A,Khomichev VV. 1992. Changes in the blood serum aminotransferase activity in the experimental infection of cercopithecus aethiops monkeys with the marburg virus. Voprosy Virusologii, 37(3): 156–157.
Subbotina E, Dadaeva A, Kachko A, Chepurnov A. 2010. Genetic factors of ebola virus virulence in guinea pigs. Virus Research, 153(1): 121–133.
Sullivan NJ, Sanchez A, Rollin PE, Yang ZY, Nabel GJ. 2000. Development of a preventive vaccine for Ebola virus infection in primates. Nature,408(6812): 605–609.
Sullivan NJ, Geisbert TW, Geisbert JB, Xu L, Yang ZY, Roederer M, Koup RA, Jahrling PB, Nabel GJ. 2003. Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature, 424(6949): 681–684.
Thi EP, Lee ACH, Geisbert JB, Ursic-Bedoya R, Agans KN, Robbins M,Deer DJ, Fenton KA, Kondratowicz AS, MacLachlan I, Geisbert TW, Mire CE. 2016. Rescue of non-human primates from advanced Sudan ebolavirus infection with lipid encapsulated siRNA. Nature Microbiology,1(10): 16142.
Warfield KL, Alves DA, Bradfute SB, Reed DK, VanTongeren S, Kalina WV,Olinger GG, Bavari S. 2007. Development of a model for marburgvirus based on severe-combined immunodeficiency mice. Virology Journal, 4: 108.
Warfield KL, Bradfute SB, Wells J, Lofts L, Cooper MT, Alves DA, Reed DK,VanTongeren SA, Mech CA, Bavari S. 2009. Development and characterization of a mouse model for marburg hemorrhagic fever. Journal of Virology, 83(13): 6404–6415.
Weingartl HM, Embury-Hyatt C, Nfon C, Leung A, Smith G, Kobinger G.2012. Transmission of Ebola virus from pigs to non-human primates.Scientific Reports, 2: 811.
Wong G, Kobinger GP. 2015. Backs against the wall: novel and existing strategies used during the 2014–2015 Ebola virus outbreak. Clinical Microbiology Reviews, 28(3): 593–601.
Wong G, He SH, Wei HY, Kroeker A, Audet J, Leung A, Cutts T, Graham J,Kobasa D, Embury-Hyatt C, Kobinger GP, Qiu XG. 2015a. Development and characterization of a guinea pig-adapted Sudan virus. Journal of Virology, 90(1): 329–399.
Wong G, Richardson JS, Cutts T, Qiu XG, Kobinger GP. 2015b.Intranasal immunization with an adenovirus vaccine protects guinea pigs from ebola virus transmission by infected animals. Antiviral Research,116: 17–19.
Wong G, Qiu XG, Richardson JS, Cutts T, Collignon B, Gren J, Aviles J,Embury-Hyatt C, Kobinger GP. 2015c. Ebola virus transmission in guinea pigs. Journal of Virology, 89(2): 1314–1323.
Wong G, Qiu XG, de La Vega MA, Fernando L, Wei HY, Bello A, Fausther-Bovendo H, Audet J, Kroeker A, Kozak R, Tran K, He SH, Tierney K, Soule G, Moffat E, Günther S, Gao GF, Strong J, Embury-Hyatt C, Kobinger G.2016. Pathogenicity comparison between the kikwit and makona ebola virus variants in rhesus macaques. Journal of Infectious Diseases, 214(S3):S281-S289.
Yamaoka S, Banadyga L, Bray M, Ebihara H. 2017. Small Animal Models for Studying Filovirus Pathogenesis. In: Current Topics in Microbiology and Immunology. Berlin, Heidelberg: Springer.
Zaki SR, Goldsmith CS 1999. Pathologic features of filovirus infections in humans. Current Topics in Microbiology and Immunology, 235: 97–116.
Zeitlin L, Whaley KJ, Olinger GG, Jacobs M, Gopal R, Qiu XG, Kobinger GP.2016. Antibody therapeutics for ebola virus disease. Current Opinion in Virology, 17: 45–49.
Zivcec M, Safronetz D, Haddock E, Feldmann H, Ebihara H. 2011.Validation of assays to monitor immune responses in the Syrian golden hamster (Mesocricetus auratus). Journal of Immunological Methods,368(1–2): 24–35.

- Zoological Research的其它文章
- Murine model of acute myocarditis and cerebral cortical neuron edema induced by coxsackievirus B4
- Parasites may exit immunocompromised northern pig-tailed macaques(Macaca leonina)infected with SIVmac239
- Development and characterization of a guinea pig model for Marburg virus
- Animal models for the study of hepatitis B virus infection
- Type I interferon receptor knockout mice as models for infection of highly pathogenic viruses with outbreak potential
- 2018 New Year Address of Zoological Research