
Molecular Simulations of FCC Dry Gas Components Adsorption in Zeolite Y1
2016-03-22 05:16DingXueLiuYibinYangChaoheShanHonghongChenFangwen
中國煉油與石油化工 2016年1期
Ding Xue; Liu Yibin; Yang Chaohe; Shan Honghong; Chen Fangwen
(1. State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266580; 2. Institute of Unconventional Hydrocarbon and New Energy Sources, China University of Petroleum, Qingdao 266580)
Molecular Simulations of FCC Dry Gas Components Adsorption in Zeolite Y1
Ding Xue1; Liu Yibin1; Yang Chaohe1; Shan Honghong1; Chen Fangwen2
(1. State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266580; 2. Institute of Unconventional Hydrocarbon and New Energy Sources, China University of Petroleum, Qingdao 266580)
Adsorption of FCC dry gas components, hydrogen (H2), nitrogen (N2), methane (CH4), ethane (C2H6) and ethylene (C2H4) in zeolite Y was studied by performing the Grant Canonical Monte Carlo (GCMC) simulations at 298 K and 823 K and under a pressure range up to 10 MPa. Simulation results were analyzed using the Langmuir model, which presented fi tting of dry gas components adsorption to be suggested as the monolayer adsorption. C2H4presented most single adsorption amount, which reached 7.63 mol/kg at 298 K under a pressure of 200 kPa. Thermodynamic parameters of the Gibbs free energy change, enthalpy change and entropy change were analyzed based on adsorption equilibrium constant obtained from the GCMC simulations. The results suggested that it was more favorable for C2H4to be adsorbed in zeolite Y. Adsorption molecules were in ordered arrangement in the zeolite, and C2H4exhibited a more orderly arrangement than other components. Additionally, a competition in the adsorption of a mixture of dry gas components was found, and supercages were the priority adsorption space. The competition was favorable to CH4and C2H6, and the competitive power was affected by temperature.
adsorption; dry gas; zeolite; molecular simulation; Monte Carlo method
1 Introduction
Zeolites are silicon aluminate crystal with abundant molecular-size pore structure and large surface area. They are widely used as adsorbents and catalysts in oil refining and chemical engineering processes[1-6], such as fluid catalytic cracking (FCC). Dry gas is the byproduct of FCC process and contains ethylene amounting to approximately 1.0 Mt/a in China[7]. However, most re fi neries have treated it as fuel to generate heat, resulting in a waste of ethylene. Therefore, the economical separation or efficient conversion of ethylene in FCC dry gas would be of significance, such as cryogenic separation[8], adsorptive separation[9]and production of ethylbenzene[10]. However, these technologies are not economical enough for most Chinese re fi neries because of scattered distribution of production units, small production scale or feed supply restriction, and in consequence these technologies are not widely applied. In our previous research, a feasible route was suggested that the ethylene from catalytic cracking dry gas could oligomerize to C3—C4hydrocarbons over FAU catalysts during the gas-solid reaction process[11].
A whole gas-solid catalytic reaction process can be divided into seven main stages, i.e. reactant external diffusion, reactant internal diffusion, surface adsorption, surface reaction, product desorption, product internal diffusion and product external diffusion. Adsorption plays an important role in this process since it can provide an effective precursor for surface adsorption. Especially for fast catalytic process, channels diffusion and surface adsorption of reactants are usually key steps that restrict the whole process. Therefore, investigation on the adsorption of FCC dry gas in FAU would be helpful to study the performance and development of oligomerization catalyst. Recently, molecular simulations have become increasingly important and popular in understanding the fundamentals of gas adsorption in zeolites at the molecular level[12-17]. Zeng, et al. simulated the adsorption ofbinary mixtures consisting of thiophene and benzene in the MFI and MOR zeolites, and demonstrated that the binary system in the MFI zeolite conformed with the competition model, while that in the MOF zeolite complied with the volume fi lling model[12]. Zhai, et al. investigated the adsorption of C4hydrocarbons in FAU, BEA and LTL zeolites by simulation and compared the adsorption isotherms, adsorbate distribution and isosteric heat of the hydrocarbons[13]. Sethia, et al. studied the sorption of several gases in ZSM-5 zeolite with different Si/Al ratios by simulation, and observed that the sorption capacity for all the gases studied decreased with an increasing Si/Al ratio of zeolite, and the effect on selectivity is in different trend for different gas[14]. Garcia, et al. investigated the adsorption isotherms of n-alkanes on Na- and Catype zeolites by using systematic molecular simulation, and con fi rmed the reality of force fi eld on predictions of adsorption[15]. Zhang, et al. performed molecular simulation of propane adsorption in FAU zeolites, and found that the increasing number of cations per unit cell could increase the adsorption loading of studied compound[16], but a reverse trend was observed in the cases of MFI zeolite which was performed by Beerdsen, et al.[17]
However, systematic studies on the adsorption capacity of zeolite for FCC dry gas are still limited. In this paper, we focus on the effects of temperature on the adsorption capacity. We present the results relating to a molecular study on adsorption of FCC dry gas components, i.e. hydrogen (H2), nitrogen (N2), methane (CH4), ethane (C2H6) and ethylene (C2H4) in the FAU-type zeolite Y. It is anticipated that this study could provide a quantitative evaluation on adsorption of dry gas components in zeolite Y, and a theoretical support for ethylene oligomerization based on FCC dry gas.
2 Simulation Details
2.1 Model construction
The FAU zeolite is a mineral group in zeolite family composed of silicate minerals. A FAU unit cell composition with a Si/Al ratio of 2.56 corresponding to 54 Al atoms per unit cell was used. It is generally defined as the zeolite Y, and is approximately equal to our experimental material. The negative charges introduced by replacing Si with Al following the Lowenstein rule are compensated by Na+in non-framework positions. The crystal structure for zeolite Y has a composition of Na54Al54Si138O384with a lattice parameter of 2.502 nm. Atom charges were chosen asqNa=+1,qSi=+2.05,qAl=+1.75,qO-Al=-1.2 andqO-Si=-1.025[17-18]. Dry gas components listed in Table 1 are constructed; the geometry optimization calculations are based on the density functional theory of Dmol3 module with generalized gradient approximation (GGA). All the calculations were performed using Materials Studio.
2.2 Method and parameters
The grand-canonical Monte Carlo (GCMC) algorithm was used in the study of adsorption problems. The interactions between guest dry gas molecules with the zeolite host framework were modeled by the Lennard-Jones (L-J) potential under CVFF force field with a cutoff radius of 1.85 nm and Coulomb potentials. The Coulombic interactions between charges in the system were calculated using the Ewald summation subject to periodic boundary conditions. The zeolite was assumed to be rigid. Based on the above method, the adsorption isotherms can be obtained directly from the simulation by computing the average number of adsorbate molecules in the zeolite framework. The non-framework cations Na+were put into simulation box randomly and allowed to move freely in the system. In order to compute the adsorption isotherms, varying reservoir pressures were applied. Simulations were carried out using the sorption module in Materials Studio. We performed simulations using one unit cell with a typical production run of 6 million Monte Carlo steps after the system reached equilibrium. The thermodynamics parameters and fugacity coef fi cients are listed in Table 1. Each component, especially the hydrocarbon, exhibits respective deviation from ideal gas at the same temperature and pressure conditions. In simulation process, pressure was modi fi ed by fugacity coef fi cient using the Peng-Robinson equation as presented below. Peng-Robinson equation:
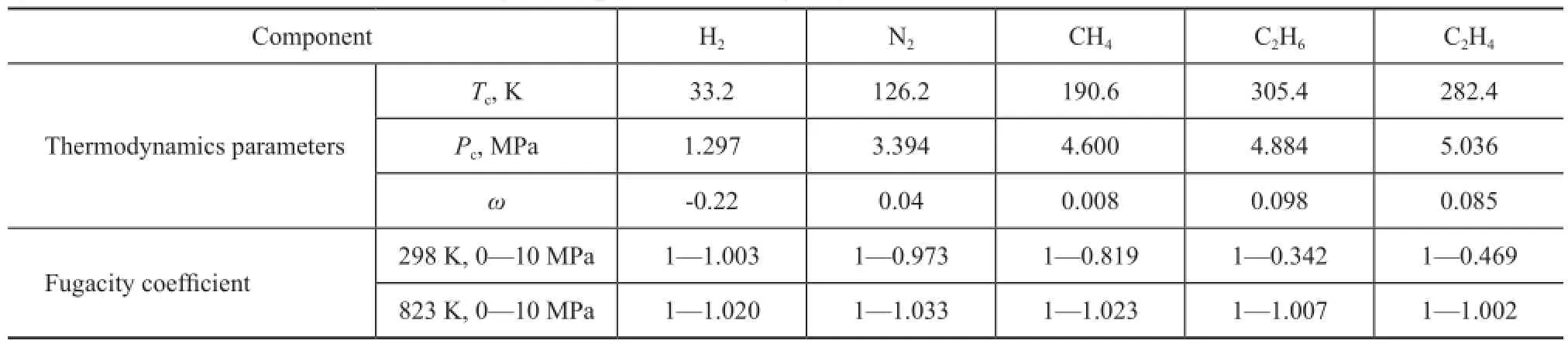
Table 1 FCC dry gas components and fugacity coefficients under various conditions
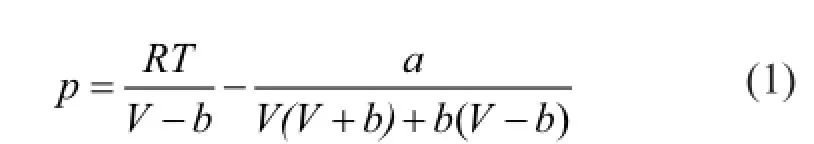
Fugacity coef fi cient equation:
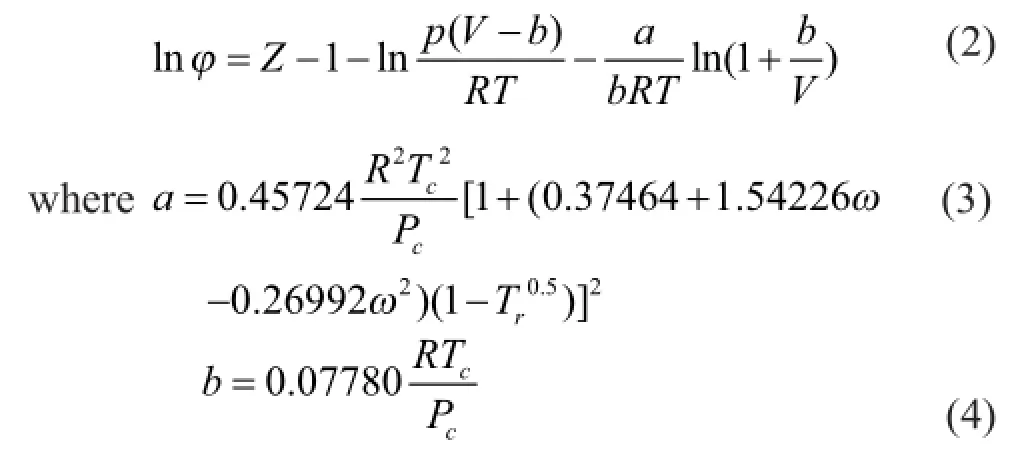
3 Results and Discussion
3.1 Single component adsorption
Adsorption isotherms were simulated for dry gas components in the FAU zeolite Y. The Langmuir model, which is used to fi t the adsorption isotherms, is given by

wherenais the absolute amount absorbed in the unit of mol/kg;nmis the maximum adsorption capacity andbis the Langmuir equilibrium constant, which represents the af fi nity between the adsorbent and adsorbate;pis the effective pressure, i.e. fugacity. The fi tting curves presented in Figures 1 and 2 show the effect of the temperature on the adsorption capacity of dry gas components under a pressure range up to 10 MPa at 298 K and 823 K, respectively. The parameters of Langmuir fittings are listed in Table 2.
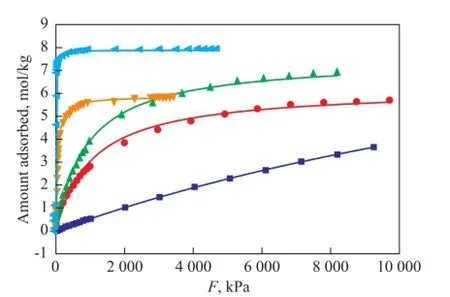
Figure 1 Single component adsorption isotherms for dry gas components at 298K under 0~10 MPa
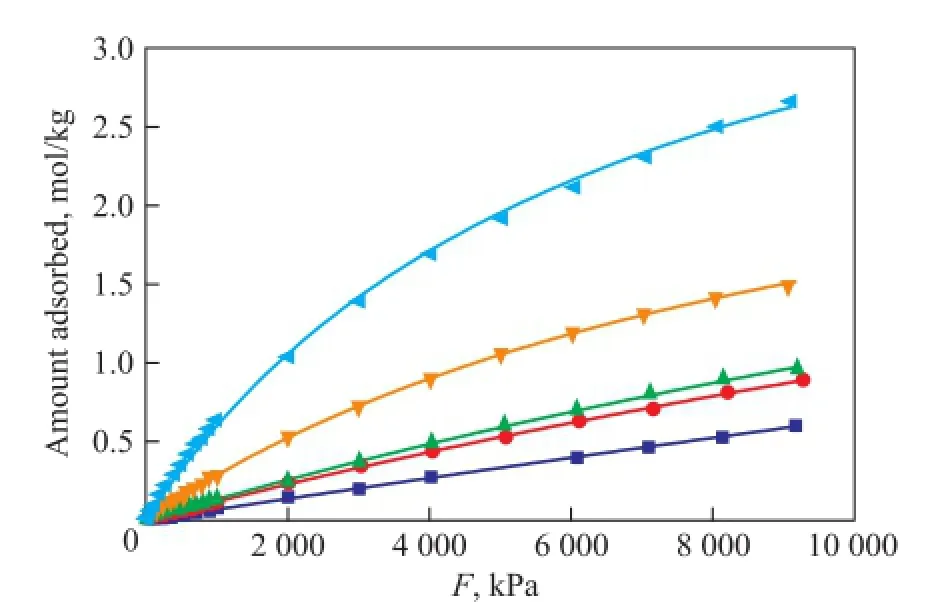
Figure 2 Single component adsorption isotherms for dry gas components at 823K under 0~10 MPa
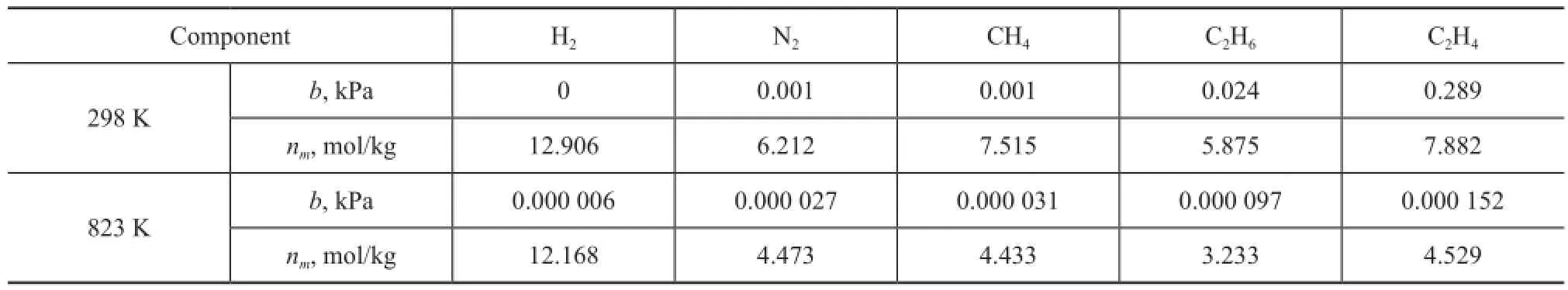
Table 2 Langmuir parameters of zeolite Y
Figure 1 indicates that the Langmuir model fi ts the data well at 298 K. All dry gas components show isotherms of type I by monolayer adsorption. H2exhibits the lowest amount adsorbed which increases linearly with the increase of pressure, and still does not reach a saturation loading under 10 MPa. N2and CH4present similar linear isotherm trend with H2at low pressure, then the growing rates slow down at high pressure, indicating an approaching saturation loading. C2H6and C2H4exhibit strongly nonlinear concave downward isotherms compared with the almost linear isotherms of H2, N2and CH4under a pressure of 400 kPa, which indicates a stronger interaction between C2H6or C2H4and the zeolite than between other molecules and the zeolite. They reach nearly the saturation loading at 1 000 kPa and 200 kPa with an adsorbed amount of 5.63 mol/kg and 7.63 mol/kg, respectively. Generally, when the pressure is below 3 500 kPa, the amount of components adsorbed varies in a decreasing order of C2H4>C2H6>CH4>N2>H2. When the adsorption of other molecules is weaker, C2H4is subject to a much stronger adsorption. When the pressure is above 3 500 kPa, CH4shows a higher adsorption amount than C2H6, but still does not reach the saturation loading at a pressure of 10 MPa (fugacity=8.19 MPa). The molecular diameter of C2H4is obviously larger than CH4, N2and H2and is approximately equal to that of C2H6, but C2H4exhibits the most amount adsorbed, while H2presents the least amount, which means that the molecular size is not the major cause of adsorption capacity, therefore the adsorption behavior of dry gas components does not fit the volume filling model[19]. Adsorption is related with intermolecular force, and it is not surprising that C2H4has π-electrons which could affect its adsorption behavior[13], moreover, the fi tting of Langmuir model for all dry gas components means that the adsorbateadsorbent interaction is stronger than the adsorbate-adsorbate interaction[20]. Based on our experimental work[7,11], adsorption at 823K was also simulated, with the results shown in Figure 2. The loading of each dry gas component decreases with an increasing temperature, but shows the similar trend as presented by Figure 1. C2H4is still the most powerful molecule. All components exhibit almost a linearly increased amount adsorbed thereby with the increase in pressure, but far from the saturation loading at 10 MPa.
3.2 Adsorption thermodynamics
According to the second law of thermodynamics, all spontaneous process is not reversible. A process is classi fi ed as spontaneous if it is characterized by a decrease in the total free energy of the system. The Gibbs free energy change is the fundamental criterion for this classi fi cation. The thermodynamic parameters of Gibbs energy change ΔG0, enthalpy change ΔH0and entropy change ΔS0for the adsorption process are calculated using the following equations.
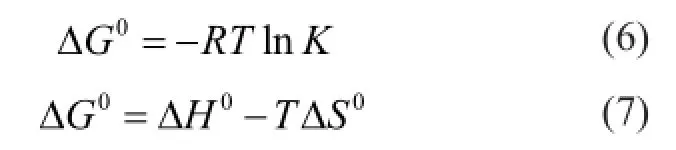
whereKis the equilibrium constant,Ris the universal gas constant (8.314 J/molK) andTis the absolute temperature (K). NormallyKis obtained from the Langmuir equation. As we have found out that the simulation data in section 3.1 indicate most components do not reach the saturated adsorption within the range of temperatures and pressures studied, thus the data are not suf fi cient for fi tting the accurate equation, and we take an alternative and more insightful approach to determineKby computingthe apparent equilibrium constantKe. It is the ratio of the concentration of adsorbate in the adsorbent which could be obtained through a separate simulation to that in the bulk phase which could be computed from Equation (1). We calculateKeat different initial concentrations of the adsorbate and then extrapolate it to zero concentration to obtain the equilibrium constantK.
Table 3 Logarithm of the equilibrium constants lnK, Gibbs free energyG0, enthalpy changeH0and entropy changeS0for the adsorption of dry gas components
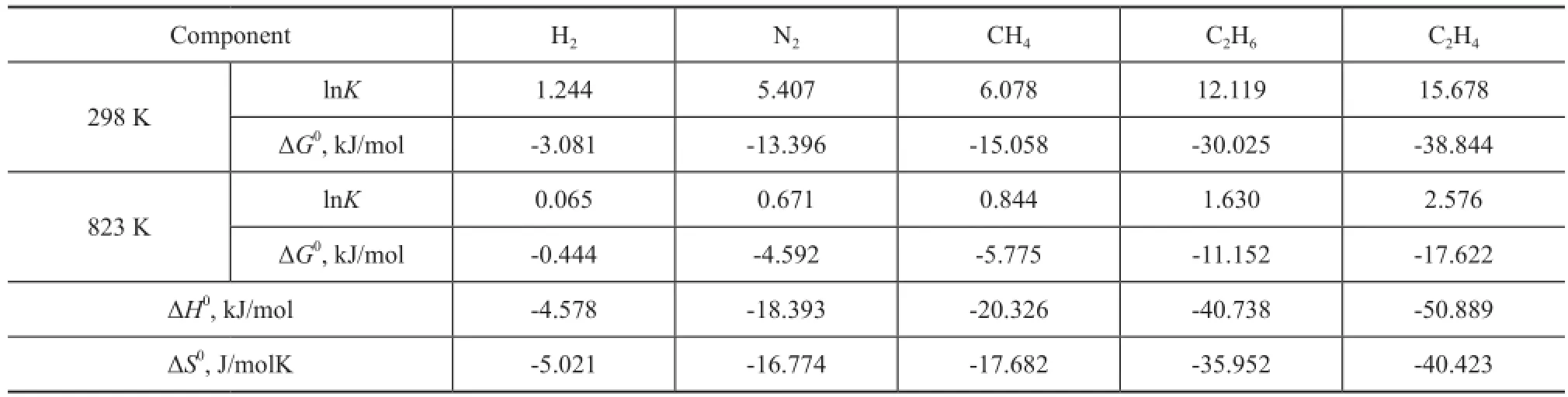
Table 3 Logarithm of the equilibrium constants lnK, Gibbs free energyG0, enthalpy changeH0and entropy changeS0for the adsorption of dry gas components
Component H2N2CH4C2H6C2H4298 K lnK1.244 5.407 6.078 12.119 15.678 ΔG0, kJ/mol -3.081 -13.396 -15.058 -30.025 -38.844 823 K lnK0.065 0.671 0.844 1.630 2.576 ΔG0, kJ/mol -0.444 -4.592 -5.775 -11.152 -17.622 ΔH0, kJ/mol -4.578 -18.393 -20.326 -40.738 -50.889 ΔS0, J/molK -5.021 -16.774 -17.682 -35.952 -40.423
In Figure 3, the apparent equilibrium constantKeis shown as a function of pressure for the adsorption of dry gas components at 298 K and 823 K, respectively. The results suggest that for each component at low pressure, the temperature dependence ofKeis evident, andKereduces significantly at 823 K compared with the case at room temperature, while the pressure effect can be ignored; however, when pressure is high enough, such as above 10 MPa, it can be speculated that the temperature dependence ofKebecomes negligible. At 298 K, for each component, with an increasing pressure,Keremains almost unchanged and then decreases obviously. Different components require different pressures to reach the turning point. The first turning point is reached at 0.001 kPa for C2H4, and then the point for C2H6, CH4, N2and H2appears successively. The equilibrium constantKis obtained by extrapolatingKeto zero pressure. The logarithm ofKand the Gibbs free energy change ΔG0calculated from Equation (6) are listed in Table 3. The results are consistent with the well-understood adsorption thermodynamics: the negative values of ΔG0suggest that the adsorption process is favorable and spontaneous for the adsorption of all dry gas components in zeolite Y at the temperatures studied; the decrease in the value of ΔG0with a decreasing temperature indicates that the adsorption process becomes more favorable at low temperatures. The comparison between each dry gas component indicates that the zeolite Y is more favorable to adsorb C2H4than other components. The enthalpy change ΔH0and entropy change ΔS0for the adsorption process can be estimated from the slope and intercept of the linear fitting relationship between ΔG0andTshown in Equation (7). The estimated results are also presented in Table 3. As listed in the table, a negative enthalpy change is observed in an increasing order of C2H4<C2H6<CH4<N2<H2, indicating that the adsorption of C2H4is more favorable than other components. The negative adsorption entropy ΔS0for all components suggests that the adsorbed dry gas molecules in the zeolite Y are in an ordered arrangement. In the adsorption process, molecules are stacked from the three-dimensional space to the two-dimensionalsurface, resulting in a decreasing randomness and motion. The more negative entropy of C2H4compared with other components re fl ects the stronger restriction on the motion of C2H4and thus shows a better performance in adsorption process. The results of Gibbs free energy, enthalpy change and entropy change are consistent, and they show that it is more favorable for C2H4to be adsorbed in zeolite Y.
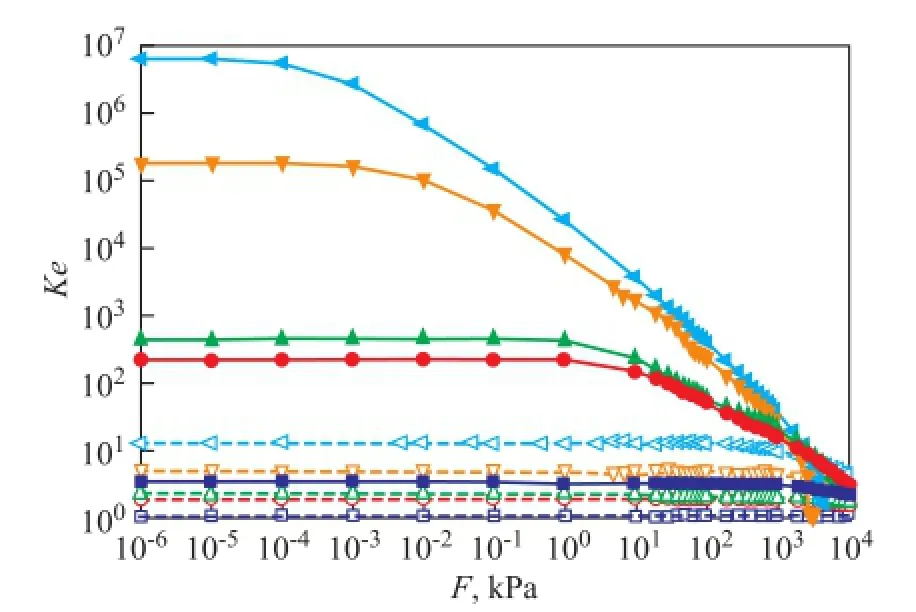
Figure 3 Apparent equilibrium constantKeas a function of pressure for the adsorption of dry gas components at 298 K and 823 K
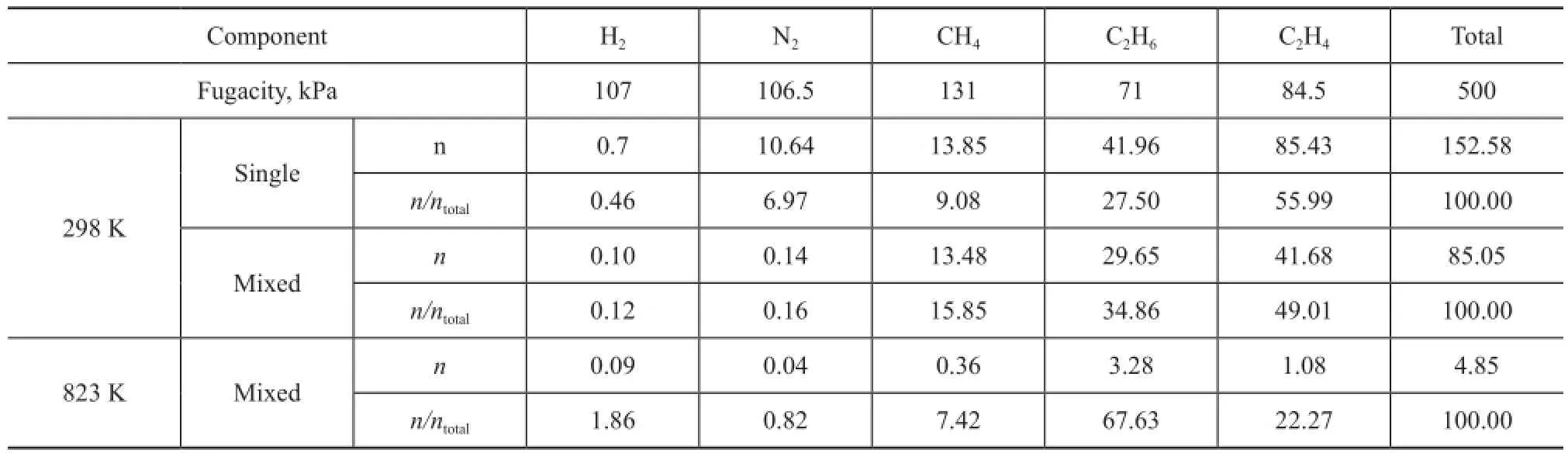
Table 4 Average loading in unit cell for dry gas components in zeolite Y
3.3 Multicomponent adsorption
Since the feed of ethylene oligomerization is a mixture, the simultaneous multicomponent adsorption for FCC dry gas is investigated. The multicomponent is obtained based on the data listed in Table 1, and the simulation pressure is equal to that in the real reaction system, i.e. 0.5 MPa. The adsorption simulation is carried out at 298 K and 823 K, respectively, and the average loadings in unit cell (n) for each component are listed in Table 4. The single component adsorption loadings for each component at 298K are also listed in the table to facilitate the data comparison.
It can be seen from Table 4 that the average loading of each mixed dry gas component absorbed thereby shows an obvious decrease compared to that of single component that is absorbed even at the same fugacity at 298 K. The total multicomponent loadings decrease to 55.7% of the total single loadings due to the restriction in adsorption space, while the effect on CH4can be ignored. Judging from the point ofn/ntotal, i.e. the loading ratio of each component versus the total, it reduces signi fi cantly for H2and N2after being mixed, while it increases for CH4and C2H6, indicating that the adsorption behavior of dry gas components can fi t the competitive adsorption model[19]. CH4exhibits the highest competitive power, while that of N2and H2is negligible. The probability density of distribution of dry gas components in the zeolite Y at 298K are presented in Figure 4. The loading of H2and N2is not included due to their negligible loading value. It can be seen from this fi gure that single CH4is mainly adsorbed in the zeolite supercages at 298 K, however, it has to contribute a part of space to other components in the multicomponent situation due to limited adsorbed position, but the adsorption is still dominant in supercages. Single C2H6occupies both supercages and channels, and it also share an adsorption position in multicomponent gas. It is more favorable for single C2H4to be adsorbed in the zeolite Y compared to other components, but C2H4is affected significantly when CH4and C2H6are present in the gas mixture. Although nearly half of the adsorption position is occupied by C2H4, nevertheless CH4and C2H6are more competitive. It can be seen from Figure 4 that the average loading of each component is reduced greatly at 823 K compared to the scenario at room temperature, moreover, the ration/ntotalchanges. C2H6occupies most of the adsorption position, which is nearly 3 times that of C2H4, indicating that the competitive power can be affected by temperature. Sufficient adsorption space provides the supercages with a most favorable position for each component. These adsorption phenomena related with the mixed components of dry gas in zeolite Y are consistent with our previous study on the adsorption process in the zeolite ZSM-5[21]。
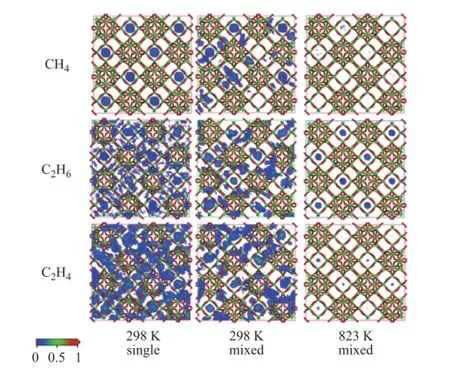
Figure 4 Probability density of distribution of dry gas components in zeolite Y
4 Conclusions
The GCMC simulation of dry gas components, i.e. H2, N2, CH4, C2H6and C2H4adsorption isotherms, was performed at 298 K and 823K and up to a pressure of 10 MPa on the zeolite Y with a Si/Al ratio of 2.56. The ad-sorption isotherms obtained from molecular simulations were fitted to the Langmuir model. Adsorption thermodynamic parameters were analyzed from the simulation results. The simulation of multicomponent adsorption was also performed. The results indicate that the Langmuir model could describe the adsorption isotherms, in which C2H4exhibited the largest adsorption amount. The thermodynamic parameters of Gibbs free energy ΔG0and enthalpy change ΔH0for the adsorption process show that the affinity between the adsorbent and adsorbate is more significant in the case of C2H4. Negative entropy change ΔS0indicates that all adsorbed molecules are in ordered arrangement in the zeolite Y. The adsorbate molecule C2H4shows a more orderly arrangement. There exists a competitive adsorption when multicomponents are adsorbed simultaneously, and the components competitiveness is affected by temperature. CH4and C2H6possess a more competitive power as compared to C2H4. It can be anticipated that although the oligomerization over the catalyst containing zeolite Y as the active component is a feasible route to make full use of the ethylene resource in FCC dry gas, and the adsorption of CH4and C2H6on catalyst surface could cause interference during reaction, so that it is bene fi cial to make the preseparation of feedstock.
Acknowledgements: We gratefully acknowledge the fi nancial support from the National Natural Science Foundation of China (No. 41302101 and No. 21476263). The authors would also like to thank Li Shuo and Yan Hao for their hard work during the course of this study.
[1] Sang Y, Jiao Q Z, Li H S, et al. HZSM-5/MCM-41 composite molecular sieves for the catalytic cracking of endothermic hydrocarbon fuel: nano-ZSM-5 zeolites as the source[J]. Journal of Nanoparticle Research, 2014, 16(12): 2755-2765
[2] De Baerdemaeker T, Yilmaz B, Muller U, et al. Catalytic applications of OSDA-free Beta zeolite[J]. Journal of Catalysis, 2013, 308: 73-81
[3] Cao Z T, Sun C J. Study on the catalyst for catalytic cracking of light gasoline to produce propylene[J]. China Petroleum Processing and Petrochemical Technology, 2010, 12 (3): 32-35
[4] Bastiani R, Lam Y L, Henriques C A, et al. Application of ferrierite zeolite in high-olefin catalytic cracking[J]. Fuel, 2013, 107: 680-687
[5] Zhao H, Song L J, Qin Y C, et al. Adsorption thermodynamics and diffusion kinetics of PX over NaY zeolite synthesized by in-situ crystallization from kaolin microsphere[J]. China Petroleum Processing and Petrochemical Technology, 2014, 16 (4): 47-54
[6] Lai J L, Song L J, Sun Z L. A frequency-response study on sorption of thiophene and benzene on NiY zeolite[J]. China Petroleum Processing and Petrochemical Technology, 2011, 13 (2): 24-28
[7] Ding X, Li C Y, Yang C H. Study on the oligomerization of ethylene in fl uidized catalytic cracking (FCC) dry gas over metal-loaded HZSM-5 catalysts[J]. Energy & Fuels, 2010, 24(7): 3760-3763
[8] Jiang G L, Xu R X, Chen H. Hydrogen and hydrocarbon separation from catalytic cracking dry gas by a membranecryogenic hybrid process[J]. Petroleum Processing and Petrochemicals, 1995, 26(1): 26-29 (in Chinese)
[9] Wang J, Ma Y J, Wang C M. Technology of producing ethylene from FCC dry gas by PSA and purification[J]. Ethylene Industry, 2006, 18(1): 60-64 (in Chinese)
[10] Chen F C, Zhu X X, Xie S J, et al. Technology development of ethylbenzene production from catalytic cracker dry-gas[J]. Chinese Journal of Catalysis, 2009, 30(8): 817-824 (in Chinese)
[11] Ding X, Li C Y, Yang C H. Oligomerization of ethylene in FCC dry gas over different solid catalysts[J]. Journal of China University of Petroleum, 2009, 33(4): 145-149 (in Chinese)
[12] Zeng Y P, Ju S G, Xing W H, et al. Adsorption of mixtures of thiophene, benzene and n-hexane in MFI and MOR using molecular simulation[J]. Acta Physico-Chimica Sinica, 2007, 23(3): 343-348 (in Chinese)
[13] Zhai D, Zhao L, Pan H F, et al. Monte Carlo investigation in C4hydrocarbon adsorption in FAU, BEA and LTL zeolites[J]. Acta Physico-Chimica Sinica, 2011, 27(6): 1400-1406 (in Chinese)
[14] Sethia G, Pillai R S, Dangi G P, et al. Sorption of methane, nitrogen, oxygen, and argon in ZSM-5 with different SiO2/ Al2O3ratios: grand canonical Monte Carlo simulation and volumetric measurements[J]. Industrial & EngineeringChemistry Research, 2010, 49(5): 2353-2362
[15] Garcia-Perez E, Dubbeldam D, Maesen T L M, et al. In fl uence of cation Na/Ca ratio on adsorption in LTA 5A: a systematic molecular simulation study of alkane chain length[J]. Journal of Physical Chemistry B, 2006, 110(47): 23968-23976
[16] Zhang J F, Burke N, Yang Y X. Molecular simulation of propane adsorption in FAU zeolites[J]. Journal of Physical Chemistry C, 2012, 116(17): 9666-9674
[17] Beerdsen E, Dubbeldam D, Smit B, et al. Simulating the effect of nonframework cations on the adsorption of alkanes in MFI-type zeolites[J]. Journal of Physical Chemistry B, 2003, 107(44): 12088-12096
[18] Calero S, Dubbeldam D, Krishna R, et al. Understanding the role of sodium during adsorption: a force field for alkanes in sodium-exchanged faujasites[J]. Journal of American Chemical Society, 2004, 126(36): 11377-11386
[19] Clark L A, Gupta A, Snurr R Q. Siting and segregation effects of simple molecules in zeolites MFI, MOR and BOG[J]. Journal of Physical Chemistry B, 1998, 102(35): 6720-6731
[20] Zhang J F, Burke N, Zhang S C, et al. Thermodynamic analysis of molecular CO2and CH4adsorption in FAU zeolites[J]. Chemical Engineering Science, 2014, 113: 54-61
[21] Ding X, Liu Y B, Yang C H, et al. Molecular simulation and thermodynamic analysis of FCC dry gas adsorption in ZSM-5 zeolite[J]. Petroleum Processing and Petrochemicals, 2015, 46(9): 58-64 (in Chinese)
Received date: 2015-11-25; Accepted date: 2015-12-23.
Professor Yang Chaohe, Telephone: +86-532-86981718; E-mail: statelab@upc.edu.cn.

- 中國煉油與石油化工的其它文章
- Experimental Study of UDS Solvents for Purifying Highly Sour Natural Gas at Industrial Side-stream Plant
- Highly Active and Stable Ni2P/SiO2Catalyst for Hydrogenation of C9Petroleum Resin
- Enhanced Performance of Denitrifying Sul fi de Removal Process by 1,2-Naphthoquinone-4-Sulphonate
- Investigation of Different Coke Samples Adhering to Cyclone Walls of a Commercial RFCC Reactor
- Numerical Study of Air Nozzles on Mild Combustion for Application to Forward Flow Furnace
- Preparation of Core-Shell Composite of Y@Mesoporous Alumina and Its Application in Heavy Oil Cracking