
The Epigenome as a therapeutic target for Parkinson’s disease
2016-02-09 05:17ShaneHegartyAideenSullivanGerardKeeffeDepartmentofAnatomyandNeuroscienceBiosciencesInstituteUniversityCollegeCorkCorkIreland
中國神經(jīng)再生研究(英文版) 2016年11期
Shane V. Hegarty, Aideen M. Sullivan, Gerard W. O’KeeffeDepartment of Anatomy and Neuroscience, Biosciences Institute, University College Cork, Cork, Ireland
The Epigenome as a therapeutic target for Parkinson’s disease
Shane V. Hegarty*,#, Aideen M. Sullivan, Gerard W. O’Keeffe*,#
Department of Anatomy and Neuroscience, Biosciences Institute, University College Cork, Cork, Ireland
How to cite this article:Hegarty SV, Sullivan AM, O’Keeffe GW (2016) The Epigenome as a therapeutic target for Parkinson’s disease. Neural Regen Res 11(11):1735-1738.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Funding:Studies in the authors’ laboratories are supported by grants from the Irish Research Council (R15897; SVH/AS/G’OK) and the National University of Ireland (R16189; SVH/AS/G’OK) and a research group from Science Foundation Ireland (SFI) under the Grant Number 15/CDA/13498 (G’OK).
Parkinson’s disease (PD) is a common, progressive neurodegenerative disease characterised by degeneration of nigrostriatal dopaminergic neurons, aggregation of α-synuclein and motor symptoms. Current dopamine-replacement strategies provide symptomatic relief, however their effectiveness wear off over time and their prolonged use leads to disabling side-effects in PD patients. There is therefore a critical need to develop new drugs and drug targets to protect dopaminergic neurons and their axons from degeneration in PD. Over recent years, there has been robust evidence generated showing that epigenetic dysregulation occurs in PD patients, and that epigenetic modulation is a promising therapeutic approach for PD. This article first discusses the present evidence implicating global, and dopaminergic neuron-specific, alterations in the methylome in PD, and the therapeutic potential of pharmacologically targeting the methylome. It then focuses on another mechanism of epigenetic regulation, histone acetylation, and describes how the histone acetyltransferase (HAT) and histone deacetylase (HDAC) enzymes that mediate this process are attractive therapeutic targets for PD. It discusses the use of activators and/or inhibitors of HDACs and HATs in models of PD, and how these approaches for the selective modulation of histone acetylation elicit neuroprotective effects. Finally, it outlines the potential of employing small molecule epigenetic modulators as neuroprotective therapies for PD, and the future research that will be required to determine and realise this therapeutic potential.
Parkinson’s disease; epigenetics; methylation; acetylation; histone acetyltransferase; histone deacetylase; small molecules
Introduction
Parkinson’s disease (PD) is a common, progressive neurodegenerative disorder, the incidence of which rises with age, with the life-time risk of developing the disease standing at 1.5% (Lees et al., 2009). The two classical hallmarks are a progressive loss of dopaminergic neurons from the substantia nigra pars compacta (SNpc), and the presence of aggregates of α-synuclein, called Lewy bodies, that are present in many regions of the central and peripheral nervous systems (Lees et al., 2009). The progressive loss of dopaminergic neurons in the SNpc leads to the motor features of the disorder which are characterised by bradykinesia, akinesia and a resting tremor. There are also many non-motor symptoms such as cognitive dysfunction, orthostatic hypotension and gastrointestinal disturbances (Lees et al., 2009). Though administration of the dopamine precursor drug, levodopa, is a successful symptomatic treatment, its effectiveness wears off over time and levodopa-induced dyskinesias develop with prolonged use. There is therefore a critical need to develop new drugs and drugs targets to protect dopaminergic neurons and their axons from degeneration in PD.
Alterations in DNA Methylation
It has become increasingly recognized that epigenetic disturbances are found in patients with PD, and that these may play a role in Parkinsonian pathology. One of the most intensively studied modes of epigenetic regulation is DNA methylation (Figure 1). This refers to the covalent methylation of residues in CpG dinucleotides within the DNA sequence, by enzymes called DNA methyltranferases (DNMTs), and most commonly results in gene repression by blocking access to DNA by transcription factors (Labbe et al., 2016). Evidence implicating global changes in the methylome is supported by observations of genome-wide changes in DNA methylation in brain and blood samples from PD patients (Masliah et al., 2013). A distinctive pattern of methylation (both increased (hyper) and decreased(hypo)) was observed in both brain and peripheral blood leukocytes from these patients, involving many genes previously associated with PD (Masliah et al., 2013). Such findings highlight the fact that methylation patterns have the potential to be employed as epigenetic biomarkers for PD. In support of these findings, a recent study examining methylation at the cellular level using induced pluripotent stem cell (iPSC)-derived dopaminergic neurons from patients with genetic (monogenic LRRK2-associated PD) and sporadic forms of PD showed extensive differences in DNA methylation, and subsequent gene expression, in iPSC-derived dopaminergic neurons from these individuals compared to controls (Fernandez-Santiago et al., 2015). Intriguingly, these differences were not seen in parental skin cells, undifferentiated iPSCs or iPSCs not enriched in dopaminergic neurons (Fernandez-Santiago et al., 2015), suggesting that there may be something unique about the dopaminergic methylome in PD. Recent animal studies have highlighted the potential for pharmacologically targeting the methylome in PD. For example, administrationof methionine (which increases global levels of DNA methylation) was found to decrease levodopa-induced dyskinesias, whereas administration of RG-108 (which reduces global levels of DNA methylation) exacerbated levodopa-induced dyskinesias (Figge et al., 2016). This highlights the importance of the DNA methylome in PD-associated motor function, and raises the potential for pharmacological manipulation of the methylome as a viable therapeutic strategy for PD.

Figure 1 Epigenetic regulation of gene expression.
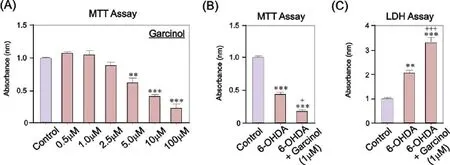
Figure 2 Garcinol dose-dependently induces cell death in SH-SY5Y cells, and significantly exacerbates the neurotoxic effects of 6-OHDA.
Alteration in Histone Acetylation
A second intensively-studied mechanism of epigenetic regulation is post-translational modification of the N-terminal tails of histone proteins, around which DNA is normally coiled (Figure 1). Although there are many types of post-translational histone modifications, including methylation, acetylation, phosphorylation, ubiquitination and sumoylation, histone acetylation at lysine residues is particularly important (Labbe et al., 2016). Histone acetylation is regulated by a balance between histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs add acetyl groups to histones, resulting in a less condensed chromatin structure, thereby facilitating transcriptional activation, whereas HDACs remove acetyl groups from histones, exerting the opposite effect (Labbe et al., 2016). As dysregulation in histone acetylation has been implicated in the pathogenesis of PD, drugs which alter levels of histone acetylation may have therapeutic potential (Harrison and Dexter, 2013). A recent study has shown that the dopaminergic neurotoxin 1-methyl-4-phenylpyridinium (MPP+) increases the levels of acetylated histones in experimental models of PD, and that there are increased levels of acetylated histones in the brains of PD patients (Park et al., 2016). This suggests that changes in the levels of histone acetylation may either 1) play a causative role in the neurodegenerative process in PD, or 2) contribute to an endogenous compensatory response in an attempt to counteract the neurodegeneration.
Potential of HDAC Inhibitors in Parkinson’s Disease
A number of studies have shown that pan- and class-specific HDAC inhibitors have neuroprotective effects in cellular models (Harrison and Dexter, 2013). However, it is important to note that not all HDAC inhibitors have been shown to be neuroprotective, with a recent study showing that a HDAC1/2 inhibitor exacerbates MPP+-induced toxicity in a SH-SY5Y cell model of PD (Park et al., 2016). Conversely, another study showed that an alternative HDAC1/2 inhibitor was neuroprotective against MPP+-induced dopaminergic toxicity bothin vitroandin vivo(Choong et al., 2016). Moreover, it has also been recently shown that other HDAC inhibitors can protect other neuronal cell types affected by PD, as it was shown that HDAC inhibition protected both dopaminergic and sympathetic neurons from MPP+-induced cytotoxicity (Collins et al., 2015). These contrasting findings are not easy to reconcile, and may reflect subtle compositional and functional differences in the HDAC inhibitor molecules. Furthermore, the precise phenotypic outcomes of HDAC inhibitors may also be concentration dependent, as highlighted by our recent work on the p300/CBP HAT described below. The potential of HDAC inhibitors for clinical translation is highlighted by an on-going Phase I clinical trial of the FDA-approved drug glycerol phenylbutyrate (an HDAC inhibitor), which is exploring the potential of this drug to increase the removal of α-synuclein from the brain (NCT02046434).
Targeting Histone Acetyltransferases in Parkinson’s Disease
In addition to HDAC inhibition, an alternative approach to increase histone acetylation is the induction of HAT activity using HAT activators. However, there has been limited research into the potential of HAT activators as potential drug therapies for PD. To begin to address this potential, we employed a selective and potent small molecular activator of p300/CBP known as CTPB (N-(4-chloro-3-trifluoromethyl-phenyl)-2-ethoxy-6-pentadecyl-benzamide) (Balasubramanyam et al., 2003). CTPB is a benzamide that activates p300/CBP HAT activity and induces p300/CBP HAT-dependent transcriptional activation, but has no effect on p300/CBP-associated factor (PCAF) or histone deacetylase activity (Balasubramanyam et al., 2003). To investigate the neurotrophic potential of CTPB in PD, we examined the survival- and growth-promoting effects of CTPB in the SHSY5Y neuronal cell line, a widely used model of human dopaminergic and sympathetic neurons (Hegarty et al., 2016). We found that CTPB-induced p300/CBP HAT activation dose-dependently promoted the survival and neurite growth of SH-SY5Y cells, and that CTPB significantly increased histone acetylation in these cells, most likely through induction of p300/CBP HAT activity. Moreover, this study found that CTPB was capable of protecting SH-SY5Y cells from the cell death induced by the dopaminergic neurotoxin 6-hydroxydopamine (6-OHDA) (Hegarty et al., 2016). Collectively these data suggest that increasing the levels of histone acetylation either through HDAC inhibition or HAT activation may be neuroprotective. However in contrast to this, another recent study found that garcinol-mediated inhibition of p300/CBP and PCAF HATs protected SH-SY5Y cells against MPP+-induced cell death (Park et al., 2016). Again, it is difficult to rationalize why both activation and inhibition of p300/CBP HATs could be neuroprotective in these cellular models of PD, but it could reflect intrinsicdifferences between CTBP and garcinol, with garcinol (but not CTPB) targeting PCAF HAT activity. Moreover, unpublished observations from our laboratory have shown that garcinol induces cell death in SH-SY5Y cells, and that it exacerbates the toxic effects of 6-OHDA (Figure 2). Further research is required to determine the basis of these contrasting findings, but again such discrepancies may reflect differences between the concentrations of garcinol used in these studies. A key challenge for future research is how to optimize the delivery of HAT-targetting molecules to the brain. Interestingly, carbon nanosphere-conjugated CTPB has the ability to cross the blood-brain barrier, localize to specific nuclei in the brain and induce hyperacetlyationin vivo(Selvi et al., 2008). Taken together, these studies demonstrate that small molecule-mediated p300/CBP HAT activation may be an avenue to explore for neurotrophic effects in PD.
Conclusion and Future Perspectives
In summary, small molecule epigenetic modulators, such as those targeting DNMTs, HDACs and HATs, hold much promise as pharmacological modifiers of the epigenetic status of the CNS, especially considering their ability to cross the blood-brain barrier. These drugs therefore have the ability to act both centrally and peripherally in the nervous system, and have the potential to protect all neurons affected by PD. However, further research is required to elucidate the precise mechanisms leading to the chronic epigenetic dysregulation observed in neurodegenerative diseases, such as PD. In addition to this, much more work is needed in translational animal models of PD for rationalizing the use of small molecule epigenetic modulators as a potential neuroprotective therapy for this disorder, including exploring strategies to deliver these drugs to the brain. Indeed, not all small molecules can cross the blood-brain barrier. As shown for CTPB, carbon nanosphere-conjugation offers a method to deliver such molecules to the brain. Although the beneficial effects of epigenetic modulators, such as HDAC inhibitors, are yet to be reported in clinical trials for PD, there is much evidence to support the continued study of molecules that target the epigenome as novel neuroprotective therapies for PD.
Author contributions:SVH wrote the manuscript, prepared the figures and edited the manuscript. AMS wrote and edited the manuscript. G’OK wrote the manuscript, prepared the figures and edited the manuscript.
Conflicts of interest:None declared.
Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK (2003) Small molecule modulators of histone acetyltransferase p300. J Biol Chem 278:19134-19140.
Choong CJ, Sasaki T, Hayakawa H, Yasuda T, Baba K, Hirata Y, Uesato S, Mochizuki H (2016) A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson’s disease. Neurobiol Aging 37:103-116.
Collins LM, Adriaanse LJ, Theratile SD, Hegarty SV, Sullivan AM, O’Keeffe GW (2015) Class-IIa histone deacetylase inhibition promotes the growth of neural processes and protects them against neurotoxic insult. Mol Neurobiol 51:1432-1442.
Fernandez-Santiago R, Carballo-Carbajal I, Castellano G, Torrent R, Richaud Y, Sanchez-Danes A, Vilarrasa-Blasi R, Sanchez-Pla A, Mosquera JL, Soriano J, Lopez-Barneo J, Canals JM, Alberch J, Raya A, Vila M, Consiglio A, Martin-Subero JI, Ezquerra M, Tolosa E (2015) Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol Med 7:1529-1546.
Figge DA, Eskow Jaunarajs KL, Standaert DG (2016) Dynamic DNA methylation regulates levodopa-induced dyskinesia. J Neurosci 36:6514-6524.
Harrison IF, Dexter DT (2013) Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease? Pharmacol Ther 140:34-52.
Hegarty SV, O’Leary E, Solger F, Stanicka J, Sullivan AM, O’Keeffe GW (2016) A small molecule activator of p300/CBP histone acetyltransferase promotes survival and neurite growth in a cellular model of Parkinson’s disease. Neurotox Res 30:510-520.
Labbe C, Lorenzo-Betancor O, Ross OA (2016) Epigenetic regulation in Parkinson’s disease. Acta Neuropathol 132:515-530.
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease. Lancet 373:2055-2066.
Masliah E, Dumaop W, Galasko D, Desplats P (2013) Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 8:1030-1038.
Park G, Tan J, Garcia G, Kang Y, Salvesen G, Zhang Z (2016) Regulation of histone acetylation by autophagy in Parkinson disease. J Biol Chem 291:3531-3540.
Selvi BR, Jagadeesan D, Suma BS, Nagashankar G, Arif M, Balasubramanyam K, Eswaramoorthy M, Kundu TK (2008) Intrinsically fluorescent carbon nanospheres as a nuclear targeting vector: delivery of membrane-impermeable molecule to modulate gene expression in vivo. Nano Lett 8:3182-3188.
*Correspondence to: Shane V. Hegarty, Ph.D. or Gerard W. O’Keeffe, Ph.D., shane.hegarty@ucc.ie or g.okeeffe@ucc.ie.
#These authors contributed equally to this work.
orcid: 0000-0003-3456-0770 (Shane V. Hegarty) 0000-0001-5149-0933 (Gerard W. O’Keeffe)
10.4103/1673-5374.194803
Accepted: 2016-10-24
- 中國神經(jīng)再生研究(英文版)的其它文章
- The status of Nrf2-based therapeutics: current perspectives and future prospects
- Targeting neuronal nitric oxide synthase as a valuable strategy for the therapy of neurological disorders
- Six psychotropics for pre-symptomatic & early Alzheimer’s (MCI), Parkinson’s, and Huntington’s disease modification
- Applicability of tooth derived stem cells in neural regeneration
- Cortical spreading depression-induced preconditioning in the brain
- Neuroinflammation, neurodegeneration and regeneration in multiple sclerosis: intercorrelated manifestations of the immune response