
Ultra-high-performance liquid chromatography for the determination of exenatide in monkey plasma by tandem quadrupole mass spectrometry
2013-12-23 06:14:52JinFengZhngChunJieShYuSunYunYunGiJiYeSunJingBinHnXinShoChunShYouXinLiWnHuiLiu
Jin-Feng Zhng, Chun-Jie Sh, Yu Sun, Yun-Yun Gi, Ji-Ye Sun,Jing-Bin Hn, Xin Sho, Chun-N Sh, You-Xin Li, Wn-Hui Liu,,*
aSchool of Pharmacy, Yantai University, Yantai 264003, PR China
bState Key Laboratory of Long-acting and Targeting Drug Delivery System, Shandong Luye Pharmaceutical Co. Ltd.,Yantai 264003, PR China
cRushan People's Hospital, Weihai 264200, PR China
1. Introduction
There has been an increasing incidence of type 2 diabetes in the world population recently. The use of traditional therapies for type 2 diabetes to achieve adequate glycaemic control was often limited by undesired hypoglycemic and weight gain. Moreover, it has been witnessed that sustained development of antidiabetic therapies exploits the enteroinsular axis. Exenatide (synthetic exendin-4), a human glucagon-like peptide-1 (GLP-1) analogue with a molecular weight of 4186.6 Da, is the first member of the class of GLP-1 receptor agonists [1-4]. It mimics the action of the human hormone GLP-1 that plays a major role in the maintenance of glucose homoeostasis [5], which could improve the glycaemic control through enhancing the glucose-dependent insulin secretion, suppressing the inappropriately elevated postprandial glucagon secretion, slowing the gastric emptying, and reducing the food intake with the result of bodyweight reducing [6,7].
Exenatide has a longer half-life (about 2.4 h) than GLP-1(less than 2 min) with no rebound effect until the termination of administration. The clinical use of exenatide still requires daily (two times a day) repeated subcutaneous injections.Recently, a slow release form of exenatide has been investigated that only requires weekly intramuscular injections,which exhibits good acceptability and compliance in clinical use [8].
To date, various methods have been developed for the quantitation of exenatide,such as immunoassay(ELISA)[8,9]and high-performance liquid chromatographic-tandem mass spectrometry (HPLC/MS/MS) method [10,11]. However, analytical ranges used in ELISA are typically limited to one to two orders of magnitude. And sometimes ELISA has poor repeatability and specificity [12]. The immunoassay methodologies also have potential interferences from endogenous compounds and require expensive antibodies and special reagents that are often difficult to obtain, which significantly lengthens the method development time and increases the cost of the experiment [11]. Compared with immunoassay methodologies, the method of UPLC/MS/MS for peptide quantitative research is potential and worthy of further exploration.The previous HPLC/MS/MS method for exenatide has a linear calibration range of 20-2000 ng/mL requiring 150 μL of blood plasma [10], although its lower limit of quantitation(LLOQ) was limited to quantitate the effective concentration of pharmacology.
Therefore, in the present study,we investigated a rapid and sensitive UPLC/MS/MS method for the determination of exenatide levels in monkey plasma and applied this method to the evaluation of Byetta after the subcutaneous injection of 5 μg for each animal. For the first time, the UPLC/MS/MS method is performed for the determination of the plasma concentration of exenatide with lower LLOQ (0.10 ng/mL),which has great future potential to be used for the monkey and human pharmacokinetics study.
2. Experimental
2.1. Reagents and chemicals
Exenatide (purity>97%) was supplied by BACHEM, Inc(Torrance, USA). Bivalirudin (purity>87%) for using as the internal standard (IS) was purchased from Shanghai TASH Biotechnology Co., Ltd. Methanol and acetonitrile are HPLC grade, purchased from Merck KGaA. Formic acid (~98%)was purchased from Fluka (Buchs, Switzerland). Ammonium hydroxide (25.0-28.0%) was purchased from LaiYang ShuangXing Chemical Co., Ltd (Shandong, China). Oasis?MCX (30 mg, 1 cc) was purchased from Waters Corporation(Milford, MA, USA). Deionized (DI) water was generated from Milli-Q water purifying system purchased from Millipore Corporation (MA, USA).
2.2. Instrumentation and chromatographic conditions
The UPLC/MS/MS system consisted of an Agilent 1290 series HPLC system (Agilent Technologies, Palo Alto, CA, USA)coupled to an Applied Biosystems Sciex Qtrap 5500 mass spectrometer (Applied Biosystems Sciex, Ontario, Canada)using electrospray ionization (ESI). Chromatography was performed on a ZORBAX RRHD Eclipse Plus C18 (1.8 μm,50 mm×2.1 mm)maintained at 40°C using a gradient elution with 0.2%formic acid as solvent A and methanol as solvent B.The gradient involved: 10% B for 1.5 min; a linear increase from 10% to 90% B in 1.0 min, 90% B for 0.5 min; a linear decrease from 90% to 10% B in 0.1 min; and equilibration at 10% B for 2.0 min. The flow rate was 0.5 mL/min without a split.
Multiple reaction monitoring (MRM) at unit resolution involved transitions of the protonated forms of exenatide at m/z 1047.4→396.3 and of bivalirudin at m/z 1090.7→650.3 in the positive ion mode. Full-scan product ion mass spectra of exenatide and bivalirudin are shown in Fig.1. Optimized MS conditions were described as follows: curtain gas, gas 1 and gas 2(all nitrogen)with 35,55 and 55 units,respectively;dwell time with 100 ms; ion spray voltage with 5500 V; source temperature with 575°C; declustering potentials with 240 V for exenatide and 100 V for bivalirudin;collision energies with 42 eV (m/z 1047.4→396.3) for exenatide and 48 eV for bivalirudin (m/z 1090.7→650.3).
2.3. Preparation of calibration standards and quality control(QC) samples
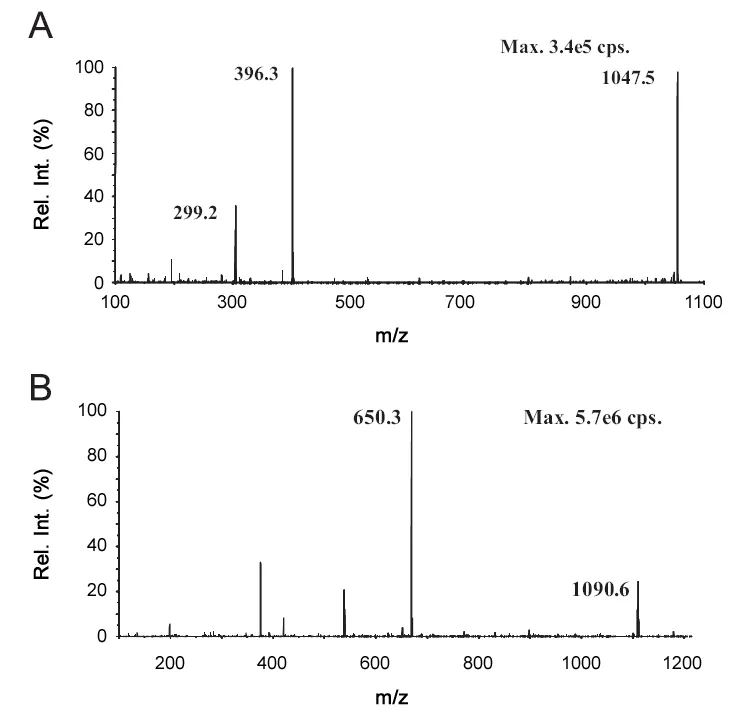
Fig.1 The product ion scans of[M+4H]4+for exenatide(A)and[M+2H]2+ for bivalirudin (B).
A stock solution of exenatide (1.0 mg/mL) in methanol(containing 0.1% formic acid) was diluted with methanol/Milli-Q water/0.1% formic acid (90:10:0.1, v/v/v) to give a series of standard solutions. A series of calibration standards were then prepared by spiking blank monkey plasma samples(50 μL) with 50 μL aliquots of standard solutions to give exenatide concentrations in plasma of 0.10,0.30,0.50,1.0,3.0,10 and 30 ng/mL, respectively. Quality control (QC) samples at three concentrations (0.20, 2.0 and 24 ng/mL) were prepared independently by the same procedure.Stock solution of the internal standard (IS, bivalirudin; 1.0 mg/mL) was also prepared in methanol (containing 0.1% formic acid).A working IS solution (bivalirudin, 8.5 ng/mL) was obtained by dilution of the bivalirudin stock solution with methanol/Milli-Q water/0.1% formic acid (90:10:0.1, v/v/v).
2.4. Sample preparation
All frozen monkey plasma samples were thawed at room temperature and subjected to SPE as follows. An aliquot of 50 μL plasma, 50 μL methanol/Milli-Q water/0.1% formic acid(90:10:0.1,v/v/v)and 50 μL IS solution were added in the 1.5 mL Eppendorf tube. The mixture was vortex-mixed for 1 min and then centrifugated at 15000×g for 5 min. After precondition of 1 mL of methanol and 0.5 mL of formic acid (0.5%) in Milli-Q water, the supernatant was transferred to the Oasis?MCX. The columns were washed with 0.5%formic acid in Milli-Q water,and then methanol/2% aqueous formic acid 96:4 (v/v). The analyte and IS were eluted with two 200 μL portions of acetonitrile/methanol/Milli-Q water/25% aqueous ammonium hydroxide(4:1:1:1.0, v/v/v/v) to 10 mL plastic tube. The collection was added with 200 μL acetonitrile/methanol/Milli-Q water/formic acid (6:5:1:0.1, v/v/v/v), and then evaporated to dryness at 50°C under a gentle stream of nitrogen.The sample was reconstituted in 50 μL methanol/water/0.1% formic acid (90:10:0.1, v/v/v) and vortexed for 30 s. A 10 μL aliquot of the sample solution was injected into the UPLC/MS/MS system.
2.5. Method validation
Assay validation was performed according to the FDA guidelines [13]. Specificity was performed by analyzing blank plasma samples from six different subjects.Three independent standard curves and six replicates of low, medium and high QC samples were analyzed on three different days. Linearity was analyzed by weighted linear regression (1/x2) of analyteinternal standard peak area ratios. The precision (represented by the relative standard deviation, RSD) and the accuracy(represented by the relative error, RE) were evaluated based on assay of the LLOQ, QC samples in three consecutive days,six replicates for each day. Recovery was determined by comparing peak areas of QC samples with those of postextraction spiked blank plasma. Matrix effects were evaluated by comparing peak areas of the analyte in QC post-extraction spiked blank plasma samples with those in standard solutions.
Stability tests were conducted to evaluate the analyte stability in stock solutions and in plasma samples under different conditions. Stock solution stability was determined by the area response of analyte (at -40°C) and internal standard(at 4°C)in the stability samples with that comparing of samples prepared from fresh stock solution. Room temperature stability(2 h),processed samples stability(reinjection stability at room temperature for 12 h), freeze-thaw stability(three cycles), long-term stability at -40°C (1 month) were performed at QC samples using three replicates at each level.Samples were considered to be stable if assay values were within the acceptable limits of accuracy (±15% SD) and precision (≤15% RSD).
2.6. Application of the method to a pharmacokinetic study in monkey
Animal studies were all performed under the instruction of protocols required by the Animal Care and Use Committee of the Chinese Academy of Medical Sciences.
The experiment was led by West China Hai-Qi Medical Technology Co., Ltd. Three male healthy monkeys (3.5-4.0 kg)were given subcutaneous injection of the same dose of Byetta(5 μg for each monkey). Blood samples (0.5 mL at each time point) were collected from the hind limbs vein prior to dosing and at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h, 12 h and 24 h after dosing. Blood samples were centrifuged and plasma was separated and stored at -40°C until analysis. The plasma concentration-time profiles for exenatide in each subject were analyzed by noncompartmental analysis using DAS 2.1.
3. Results and discussion
3.1. Mass spectrometry
The [M+4H]4+of exenatide was selected as the precursor ion(m/z 1047.4) because it was the principal charged species observed in the Q1 scan process. The corresponding[M+2H]2+of bivalirudin (m/z 1090.7) was chosen for the same reason. Accordingly, the transition m/z 1047.4→396.3 was selected as quantifier. The transition m/z 1090.7→650.3 was used to bivalirudin.
3.2. Method development
Various mobile phase combinations of acetonitrile, methanol,ammonium acetate and formic acid were investigated to optimize the chromatography for sensitivity, speed and peak shape.Methanol gave a better response than acetonitrile and the peak shape was improved by the addition of formic acid. The ZORBAX RRHD Eclipse Plus C18(1.8 μm, 50 mm×2.1 mm)was chosen because it gave the best resolution and peak shapes with minimal matrix effects.At the same time,we found that the dilution of exenatide had a great effect on the signal intensity and that the acidity of the dilution gave great contribution to the stability of exenatide. The dilution was finally confirmed as the methanol/Milli-Q water/0.1% formic acid (90:10:0.1, v/v/v).
Isolation of exenatide from plasma is a tedious task,because of the low concentration levels and interferences induced by matrix. The result of protein precipitation was not comforting [14], which should be related to the relatively big size of the peptide drug that leads to the co-precipitation of exenatide. The recovery of reverse phase liquid-liquid extraction was low, which might be due to the related properties of amphoteric compound.According to the information,various solid-phase extractions (Waters Oasis?HLB, Waters Oasis?MCX, Waters Oasis?WAX) were investigated. Sample prepared by HLB and WAX showed the poor recovery.Oasis?MCX with appropriate condition of elution provided better recovery and less remaining endogenous interferences.The percentage of formic and aqueous ammonium in the elutions was a key factor.Glass tube and plastic tube were also investigated as the receiving vessel, and the results indicated that the glass tube had high adsorption of exenatide.It should be suggested that exenatide contained more acidic amino acids leading to the more easily glass adsorption [15,16].
A number of potential internal standards including triptorelin, elcatonin, and diazepam were evaluated in the present study, while most of them were incompatible with the sample preparation procedure because of their physicochemical properties.Finally,bivalirudin was chosen for the suitable IS for its similar physicochemical characteristics and similar retention time. It could be presumed that bivalirudin and exenatide had more acidic amino acid than basic amino acid in their structures while some others went in the opposite direction.Moreover, the results of our study showed that the pH of compounds was an important factor in the complex elution systems of MCX column.
Blank plasma of several animals including rat,dog,monkey and human were compared; however, there was no interference with the endogenous substances only in the blank monkey and human plasma as shown in Fig.2, which might be caused by the different kinds and contents of GLP-1 and its analogues in various species plasma.
3.3. Method validation
3.3.1. Selectivity and chromatography
The assay was found to be highly specific since there were no endogenous peaks in monkey plasma at the retention times of IS and exenatide. Representative chromatograms of blank plasma and blank plasma spiked with exenatide at the LLOQ(0.10 ng/mL) were shown in Fig.3.
3.3.2. Linearity
Calibration curves were prepared using seven standards,and it was linear in the range of 0.10-30 ng/mL. The mean correlation coefficient (r2) was greater than 0.9986 for all curves.
3.3.3. Precision and accuracy
As shown in Table 1, the precision and accuracy of exenatide in the intra- and inter-day runs were within ±15% at LOQ,MOQ, and HOQ concentrations and within ±20% at LLOQ QCs.
3.3.4. Extraction efficiency
The recoveries of exenatide and the IS were good and reproducible. Recoveries of exenatide from the low, medium and high QC samples were 65±2%, 63±3% and 63 ±3%, respectively. The recovery of bivalirudin was 69 ± 4%.
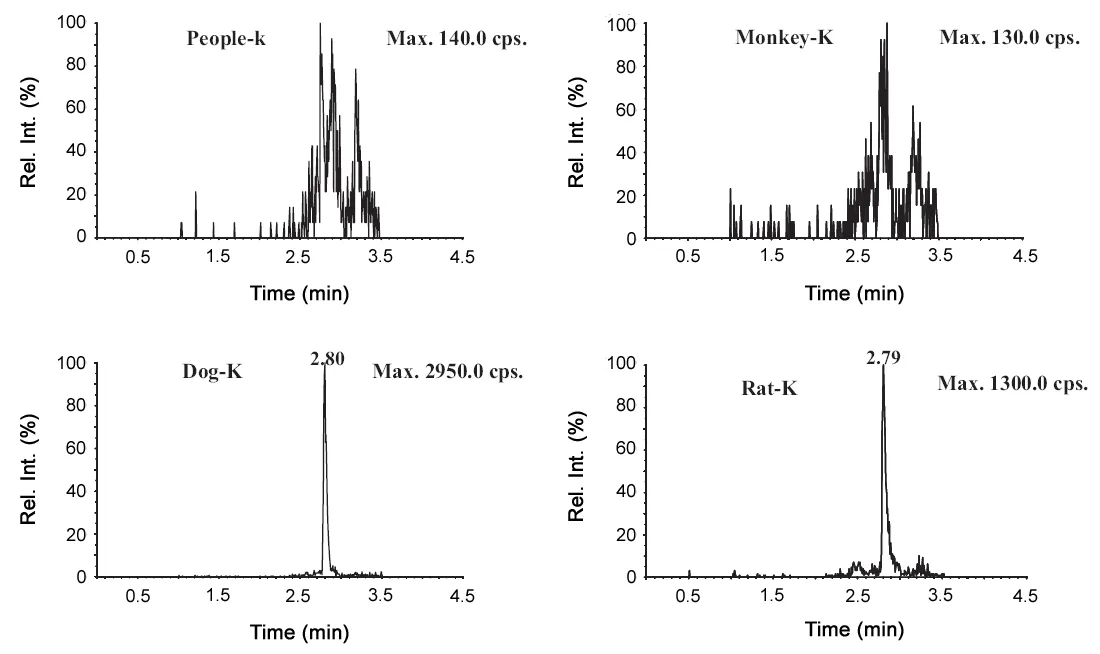
Fig.2 Representative MRM chromatograms of exenatide(m/z 1047.4→396.3)in four different animals blank plasma(human,monkey,dog and rat).
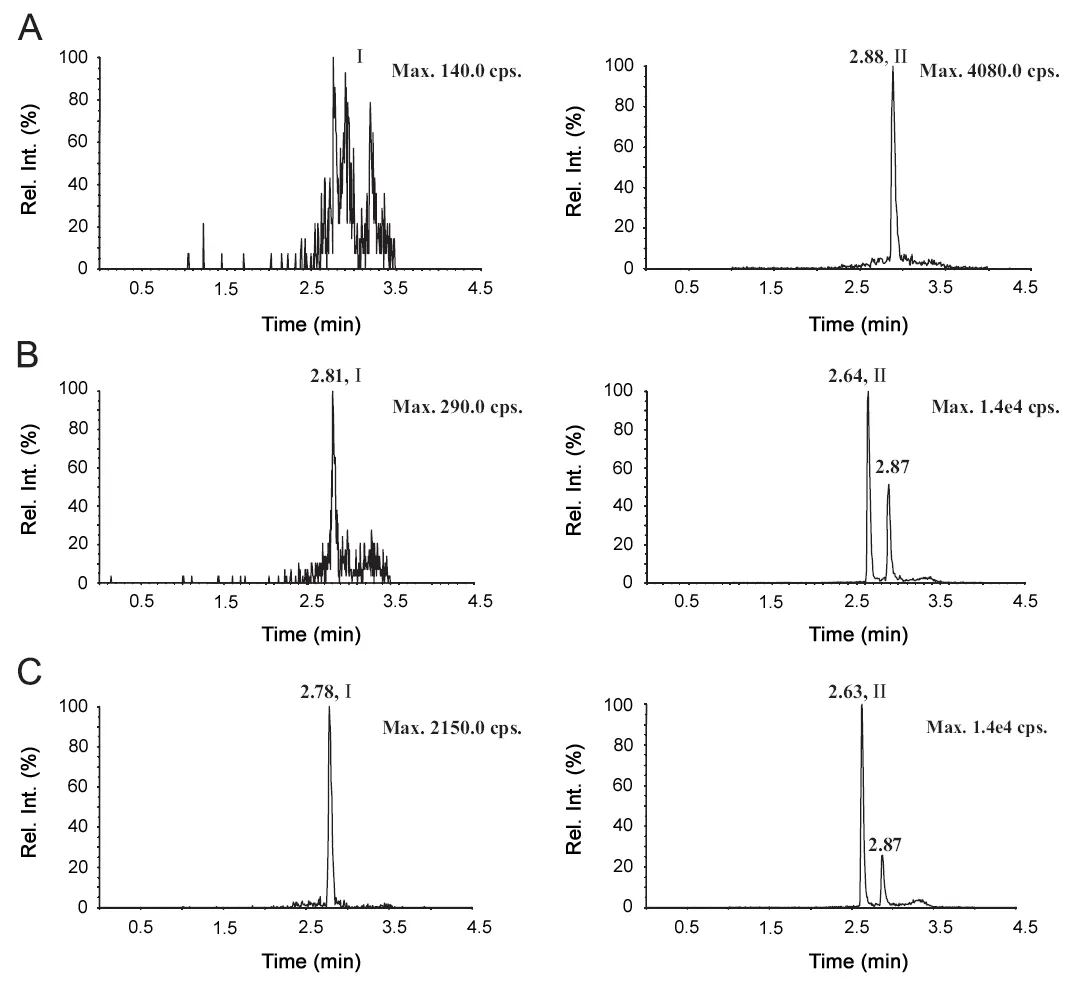
Fig.3 Representative MRM chromatograms of exenatide and bivalirudin in monkey plasma. (A) Blank plasma; (B) blank plasma spiked with 0.1 ng/mL of exenatide (I) and 8.5 ng/mL bivalirudin (II); (C) plasma sample from a monkey volunteer 15 min of exenatide after the subcutaneous injection of exenatide (5 μg for each monkey).
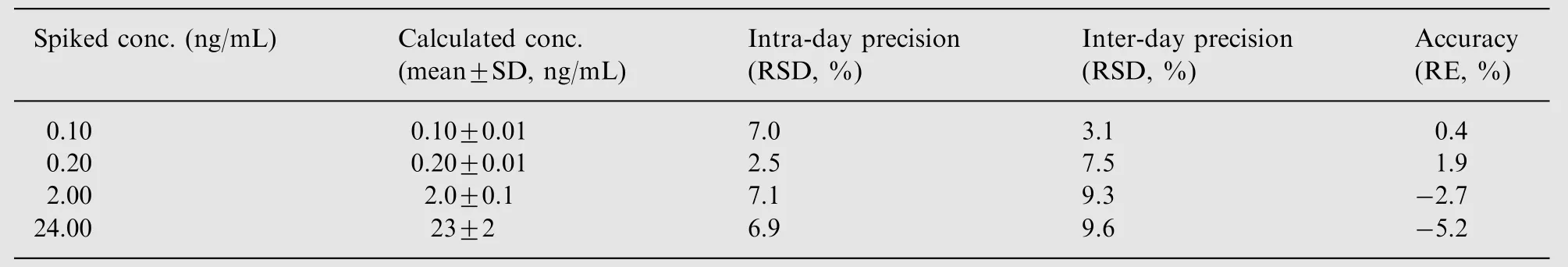
Table 1 Precision and accuracy for the determination of exenatide in monkey plasma (data are based on assay of six replicates on three different days).
3.3.5. Matrix effect
The matrix effects were shown to be minimal based on the ratios of the peak areas for exenatide in the low, medium and high post-extraction spiked blank plasma samples to the peak areas in the corresponding standard solutions; these ratios were 106±4%, 104±7% and 105±2%,respectively. Similarly the ratio of bivalirudin was 105± 8%.
3.3.6. Stability
The stock solutions of exenatide were stable at -40°C for at least one month and IS solutions were stable at 4°C for at least three months.
Room temperature stability, reinjection stability, freezethaw stability and long-term stability for exenatide were investigated at the low, medium and high QC samples. The results revealed that exenatide was stable under all of the conditions examined (Table 2).
3.4. Pharmacokinetic study
The mean concentration-time profile of exenatide after the subcutaneous injection of exenatide is shown in Fig.4. The mean peak plasma concentration (Cmax) was 2.1±0.4 ng/mL at a time(Tmax)of 0.5 h after the injection was given.The mean area under the plasma concentration-time curve (AUC0-t) was 5.1±0.7 ng ·h/mL and the mean elimination half-life(t1/2)was 1.2±0.2h.
4. Conclusion
In summary, a highly sensitive and selective UPLC/MS/MS method was first developed for the determination of exenatide in monkey plasma, which had the LLOQ of 0.10 ng/mL and good accuracy and precision. The lower requirement ofmonkey plasma volume was only 50 μL, which provided the possibility for repeated analyses without increasing blood volumes. Because there was also no interference with the endogenous substances in human plasma, this method could be well applicable to the clinical pharmacokinetic research.
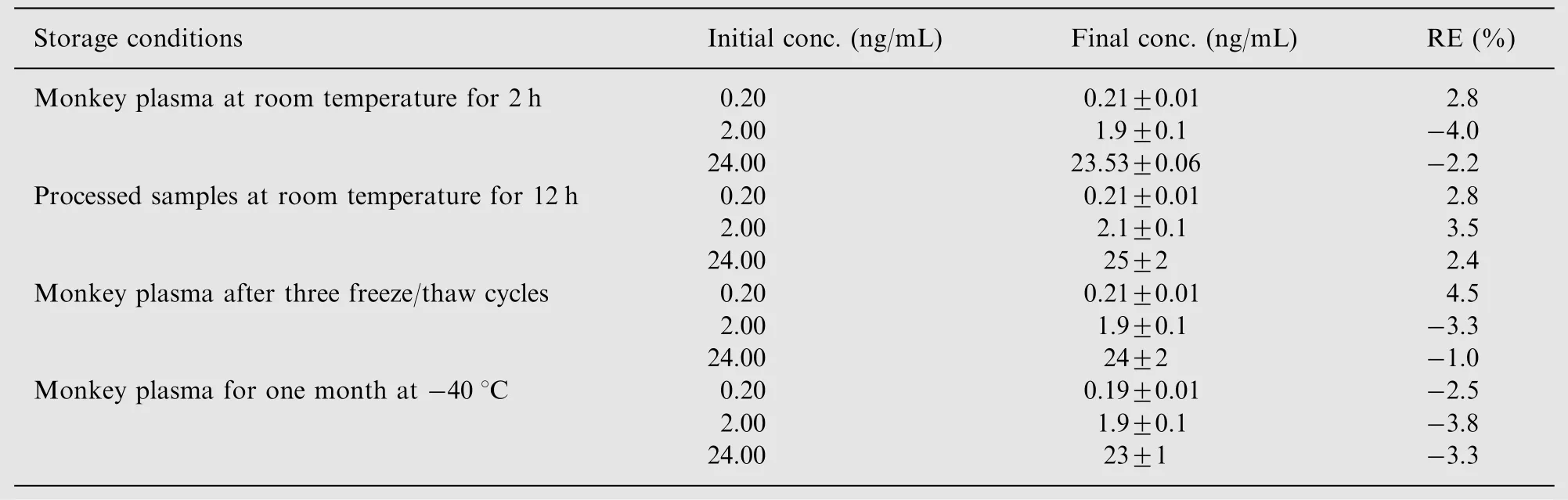
Table 2 Stability of exenatide under various conditions (data are mean±SD of three replicates).
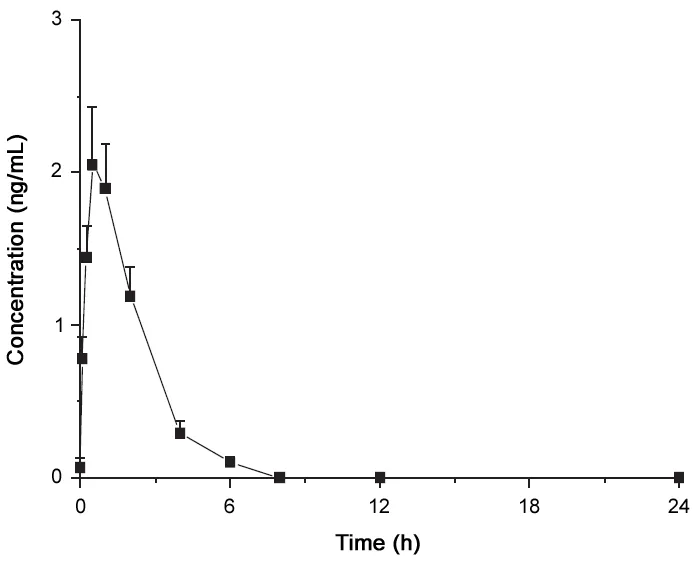
Fig.4 Plasma concentration-time profile of subcutaneous injection of exenatide(5 μg for each monkey)to healthy monkey.Data are mean±SD for three monkeys (three males).
This work was supported by the National Basic Research Program of China (973 Program) (No. 2010CB735602 and No. 2012CB724003). We also would like to thank Taishan Scholar Projects for their kindly supported in the method development.
[1] P.R. Flatt, C.J. Bailey, B.D. Green, Recent advances in antidiabetic drug therapies targeting the enteroinsular axis, Curr. Drug Metab. 10 (2009) 125-137.
[2] B.T. Srinivasan, J. Jarvis, K. Khunti, et al., Recent advances in the management of type 2 diabetes mellitus: a review, Postgrad.Med. J. 84 (2008) 525-526.
[3] J.S. Freeman, A physiologic and pharmacological basis for implementation of incretin hormones in the treatment of type 2 diabetes mellitus, Mayo. Clin. Proc. 85 (12) (2010) 7-9.
[4] V.R. Aroda, R. Ratner, The safety and tolerability of GLP-1 receptor agonists in the treatment of type 2 diabetes: a review,Diabetes Metab. Res. Rev 27 (2011) 528-529.
[5] R. Jones, The safety and tolerability of GLP-1 receptor agonists in the treatment of type-2 diabetes, Int. J. Clin. Pract. 64 (2010)1403-1414.
[6] P.A. Kothare, H. Linnebjerg, Y. Isaka, et al., Pharmacokinetics,pharmacodynamics, tolerability, and safety of exenatide in Japanese patients with type 2 diabetes mellitus, J. Clin. Pharmacol. 48 (2008) 1390.
[7] D.P. Bradley, R. Kulstad, D.A. Schoeller, Exenatide and weight loss, Nutrition 26 (2010) 245-247.
[8] M. Fineman, S. Flanagan, K. Taylor, et al., Pharmacokinetics and pharmacodynamics of exenatide extended-release after single and multiple dosing, Clin. Pharmacokinet. 50 (1) (2011) 69-72.
[9] K. Kristen M, The current status of exenatide once weekly, Clin.Med. Insights: Ther. 3 (2011) 223-224.
[10] J.R.Kehler,C.L.Bowen,S.L.Boram,et al.,Application of DBS for quantitative assessment of the peptide Exendin-4;comparison of plasma and DBS method by UPLC-MS/MS, Bioanalysis 2 (8)(2010) 1461-1464.
[11] L. Dillen, W. Cools, L. Vereyken, et al., Comparison of triple quadrupole and high-resolution TOF-MS for quantification of peptides, Bioanalysis 4 (5) (2012) 566-575.
[12] A.N. Hoofnaqle, M.H. Wener, The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry, J. Immunol. Methods 34 (1-2) (2009) 3-11.
[13] Guidance for Industry Bioanalytical Method Validation,U.S.Departmentof Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research(CDER), Center for Veterinary Medicine(CVM), BP, May 2001〈http://www.fda.gov/cder/guidance/4252fnl.htm〉.
[14] R.W.Zhang,W.T.Liu,L.L.Geng,et al.,Quantitative analysis of a novel antimicrobial peptide in rat plasma by ultra performance liquid chromatography-tandem mass spectrometry, J. Pharm.Anal. 1 (3) (2011) 192.
[15] I.D.V. Broek, R.W. Sparidans, J.H.M. Schellens, et al., Quantitative bioanalysis of peptides by liquid chromatography coupled to(tandem)mass spectrometry,J.Chromatogr.B 872(2008)1-7.
[16] K. Heining, T. Wirz, Determination of taspoglutide in human and animal plasma using liquid chromatography-tandem mass spectrometry with orthogonal column-switching, Anal. Chem. 81(2009) 3705-3711.
 Journal of Pharmaceutical Analysis2013年4期
Journal of Pharmaceutical Analysis2013年4期
- Journal of Pharmaceutical Analysis的其它文章
- LC-MS/MS determination and pharmacokinetic study of bergenin, the main bioactive component of Bergenia purpurascens after oral administration in rats
- Characterization of phloroglucinol derivatives and diterpenes in Euphorbia ebracteolata Hayata by utilizing ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry
- Application of analytical instruments in pharmaceutical analysis
- Investigation of the interaction between indigotin and two serum albumins by spectroscopic approaches
- In vitro antibacterial and free radical scavenging activity of green hull of Juglans regia
- Fingerprint analysis of Cirsium japonicum DC. using high performance liquid chromatography